

1 Introduction
In the last two decades, important progress has been achieved in the development of rational strategies to control the structures and metal composition of multinuclear transition complexes. Part of this interest is due to the presence of multimetallic coordination sites that could facilitate unique chemical transformations through cooperative or successive interaction of the metal centres with substrate molecules [1–2].
Complexes having two widely divergent metals, on the one hand, a Lewis-acid early transition metal and, on the other one, an electron-rich late transition metal, are promising candidates for new stoichiometric and catalytic reactions. Moreover, they could model the role of the support in heterogeneously catalysed reactions [3–4]. If the metal centres could “speak each other”, properties such as multifunctionality and cooperative effects should be expected. Nevertheless, the problems found to meet the different coordination environments of both metals in close proximity could be as great as the expectations, and thus synthetic strategies leading to new complexes are highly desirable. A goal is still to find ways to accommodate the different electronic and coordination environments required by the metals in close proximity, in order to obtain stable compounds.
Transition-metal complexes with bridging sulphur ligands have a broad scope comprising from a widely studied biological systems and applied industrial processes such as hydrodesulphurisation and catalysis to novel chemistry of molecular systems [5–8]. Organometallic early-late heterobimetallic complexes are relatively uncommon and efforts to develop this chemistry have been reviewed recently [9]. In particular, an elegant synthetic approach for early-late heterobimetallic complexes established by Stephan et al., and other groups, involves early-metal complexes acting as metalloligands for late transition metal species [10]. They include titanium and zirconium complexes with terminal thiolate [11], phosphide [12], and alkoxyalkylphosphine ligands [13], pendant chelating metalloligands [14–15] and also macrocyclic metalloligands [16]. Other useful metalloligands are based on early-metallocene complexes [17] and remarkable synthetic strategies involve the use of bis (cyclopentadienyl) titanacyclobutanes [18–20] or of methylcarbene titanium complexes [21] to give methylene bridged early-late heterometallic complexes.
We report in this account recent progress on the preparation and reactivity of early – titanium and zirconium –, late – rhodium and iridium – transition metals sulphido complexes. The effectiveness of sulphur is well known to behave as a good bridging ligand preventing the undesirable fragmentation of the heteronuclear framework. Our synthetic approach was based on the deprotonation of the bis-hydrosulphido titanium or zirconium complexes [Cp2M(SH)2] (M = Ti, Zr) by appropriate d8 rhodium and iridium compounds containing diolefins or carbonyl groups as ancillary ligands. At first glance, the preparation of heterotrinuclear complexes containing the ‘Cp2M(μ-S)2M′2’ (M′ = Rh, Ir) framework should be expected, but this behaviour is only favoured for zirconium, while the loss of one coordinated cyclopentadienyl ligand from the titanium centre is dominant. This loss creates additional coordination vacancies that should facilitate the preparation of complexes of higher nuclearity. We herein report on this rich heterometallic chemistry involving titanium and zirconium as early metals, and rhodium and iridium, as late transition metals.
2 Sulphido-bridged titanium–rhodium and titanium–iridium complexes
2.1 Heterobimetallic TiRh3 and TiIr3 diolefin complexes
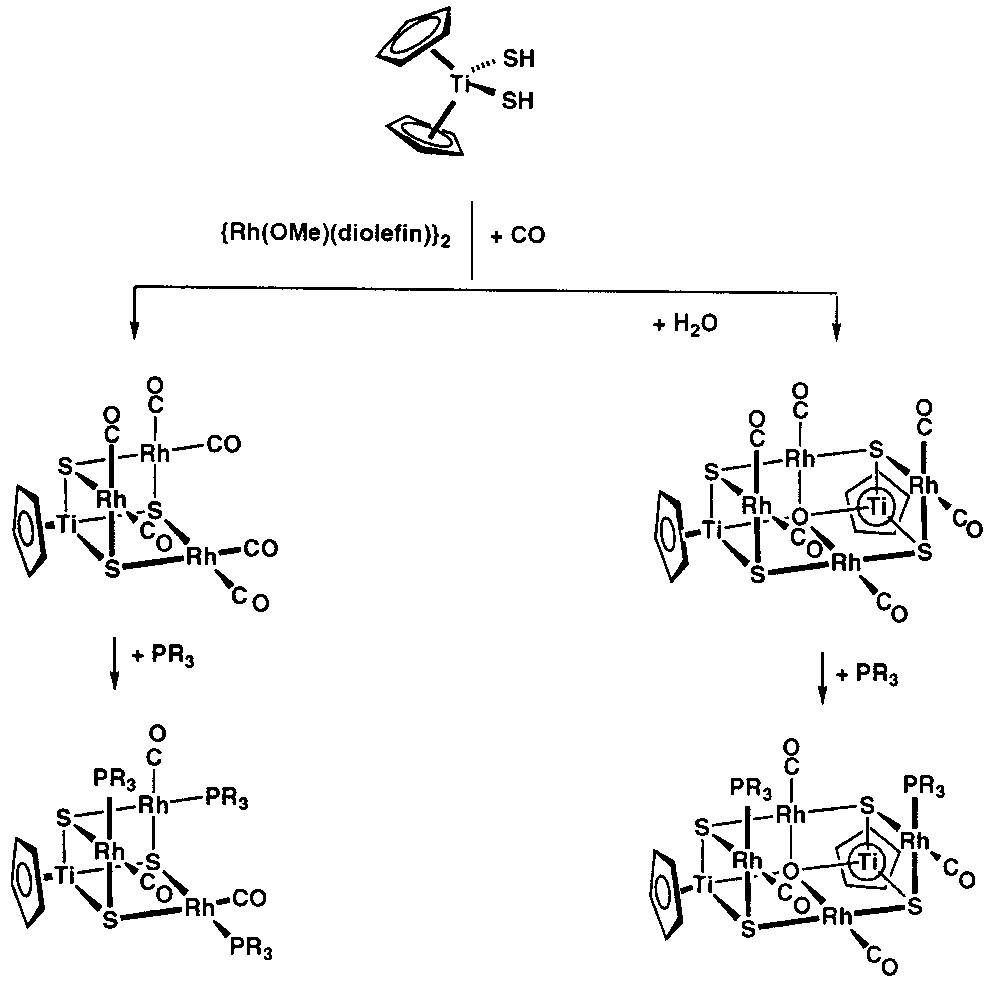
The reported reactivity of [Cp2Ti(SH)2] is very limited since its preparation in 1965 [22]. We initially recognised the ability of this compound to stabilise early-late polynuclear complexes by protonation of appropriate late transition metal complexes. We initially thought that the protonation of [{Rh(μ-OMe)(diolefin)}2] (diolefin = 1,5-cyclooctadiene, cod; tetrafluorobenzobarrelene, tfbb) by [Cp2Ti(SH)2] would yield heterotrinuclear complexes of the type [Cp2Ti(μ3-S)2{Rh(diolefin)}2], but the reaction was more complicated. Monitoring the reaction by 1H NMR spectroscopy, we observed that it proceeds quickly with formation of methanol, free cyclopentadiene and heterotetranuclear complexes of formula [CpTi(μ3-S)3{Rh(diolefin)}3] as main products [23–24] (Fig. 1).
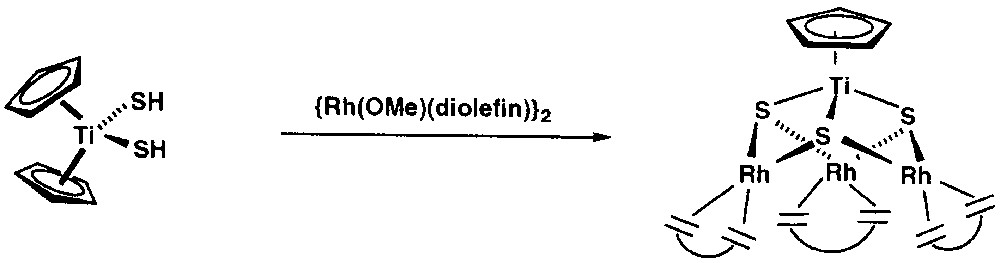
Synthesis of [CpTi(μ3-S)3{Rh(diolefin)}3] from [Cp2Ti(SH)2].
A similar protocol using [Cp2Ti(SH)2] and [{Ir(μ-OMe)(cod)}2] was not suitable for the synthesis of the related Ti–Ir complex [CpTi(μ3-S)3{Ir(cod)}3], since this reaction followed an alternative pathway resulting in the formation of the known cluster [Ir3(μ-S)2(μ-H)(cod)3] in high yield [25]. As the latter hydridoiridium cluster can be obtained from [{Ir(μ-Cl)(cod)}2] and NaHS or H2S, the titanium complex acts, in fact, as a source of hydrosulphido ligands for iridium, one of which produces the oxidative addition of the S–H bond, leading to the hydrido ligand. To avoid this undesired reaction, we decided to use a deprotonated form of [Cp2Ti(SH)2]. Thus, [Cp2Ti(SH)2] was reacted with butyllithium, yielding a green solution of the anionic complex [Cp2Ti2(μ-S)2(S)2]2–, that showed identical colour and 1H NMR spectrum than that for the Na2[Cp2Ti2(μ-S)2(S)2] complex, recently isolated by Kubas [26] from the monodeprotonation of Cp2Ti(SH)2 with sodium hydride. Solutions of the anion [Cp2Ti2(μ-S)2(S)2]2–, prepared in situ, were excellent starting materials for the synthesis of heterometallic iridium–titanium and rhodium–titanium complexes with sulphido bridging ligands providing also an insight on the above reactions. On reacting [Cp2Ti2(μ-S)2(S)2]2– with the chloro-complexes [{M(μ-Cl)(diolefin)}2], the compounds [CpTi(μ3-S)3{M(diolefin)}3] (M = Rh, diolefin = cod, nbd, tfbb; Ir, diolefin = cod) resulted [23]. The complexes were isolated as air-stable crystalline solids. Their NMR spectra were consistent with the incomplete cubane structure lacking one vertex, in which the group [CpTi(S)3]3– capped a trimetallic triangle of the late metals, as substantiated for [CpTi(μ3-S)3{Rh(tfbb)}3] by X-ray diffraction methods [24]. This tetrafluorobenzobarrelene complex presented one titanium and three rhodium atoms engaged through three alternated triple-bridging sulphur atoms, each connecting two rhodium atoms to the titanium centre. The central TiRh3S3 core exhibited an incomplete cubane-type structure, with one vacant site close to the three rhodium atoms, and the internal bond angles deviated slightly from the ideal value of 90°. The Rh–Ti separations were in the range 2.912–2.940(3) Å. These distances and the values of the Ti–S–Rh angles are consistent with a week dative interaction from the electron rich d8 metal to the acid d0 titanium centre. A related oxoderivative of formula [Cp*Ti(μ-O)3{Rh(cod)}3] was simultaneously reported, but in this case the titanium source was specifically a monocyclopentadienyl compound of formula [Cp*TiMe3] [27].
The reactions leading to [CpTi(μ3-S)3{Rh(diolefin)}3] complexes from [Cp2Ti(SH)2] with the methoxorhodium complexes should proceed through a complicated mechanism sensitive to nucleophiles, such as methanol, produced in the reactions. Remarkable observations on these syntheses were the incorporation of an extra sulphido ligand in the products, the removal of a cyclopentadienyl ligand initially bonded to titanium and the loss of some titanium from the starting material as unknown cyclopentadienyl products (Fig. 2). The transfer of a sulphido ligand between two titanium atoms requires the intermediacy of species with sulphido ligands bridging two titanium centres. The formation of such dinuclear species and the removal of the cyclopentadienyl ligand in these reactions was exemplified in a simple way by the reaction of Cp2Ti(SH)2 with BuLi. For this simplest case, monodeprotonation of the titanium complex, either with BuLi or NaH, should take place in a first instance to give the undetected intermediate [Cp2Ti(S)(SH)]–, followed by a transfer of the proton on the hydrosulphido ligand to one cyclopentadienyl ring. Similarly, [Cp2Ti(SH)2] should be deprotonated by [{Rh(μ-OMe)(cod)}2] to give methanol and the dinuclear complex [Cp2Ti(μ-S)(μ-SH)Rh(diolefin)], A. This unobserved intermediary species would lose the cyclopentadienyl ring, as does the anion [Cp2Ti(S)(SH)]–, either spontaneously or through interaction with methanol. Removal of one Cp ring from the reaction of bis(cyclopentadienyl) titanium complexes with methanol is a well-known feature of this kind of compounds [28]. In any case, the dimerisation of the resulting species would afford tetranuclear species of formula [(CpTi)2(μ3-S)4{Rh(diolefin)}2], B, with four sulphido bridging ligands. Complex B is also the expected product from the reactions of the anionic complex [Cp2Ti2(μ-S)2(S)2]2– with the chlorocomplexes [{M(Cl)(diolefin)}2]. Thus, both synthetic routes have in common this intermediate B, which possesses a cubane-type sulphido cluster structure. A further reaction of complex B with [{M(μ-Cl)(diolefin)}2] or [{M(μ-OMe)(diolefin)}2] would lead to the asymmetrical breaking of the sulphido bridges rendering the heterotetranuclear complexes [CpTi(μ3-S)3{M(diolefin)}3] along with unknown cyclopentadienyltitanium complexes. This simplified mechanism explains the features for both synthetic approaches assuming the simplest intermediates. In particular, the proposed intermediate species, B, of formula [(CpTi)2(μ3-S)4{Rh(cod)}2] was isolated by reacting the thiotitanate anion, [Cp2Ti2(μ-S)2(S)2]2–, with [{Rh(μ-Cl)(cod)}2] [29]. The synthesis and molecular structures of cubane-type sulphido clusters have been reported by Hidai et al. [29–30], that have confirmed our previous proposal of formation of [CpTi(μ3-S)3Rh3(diolefin)3] by the intermediacy of [(CpTi)2(μ3-S)4{Rh(diolefin)}2] and [Cp2Ti2(μ-S)2(S)2]2– compounds, according to Fig. 2. In this context, it is important to mention the development, by Hidai et al., of a variety of rational synthetic approaches for the preparation of cubane type sulphido clusters containing noble metals [30].
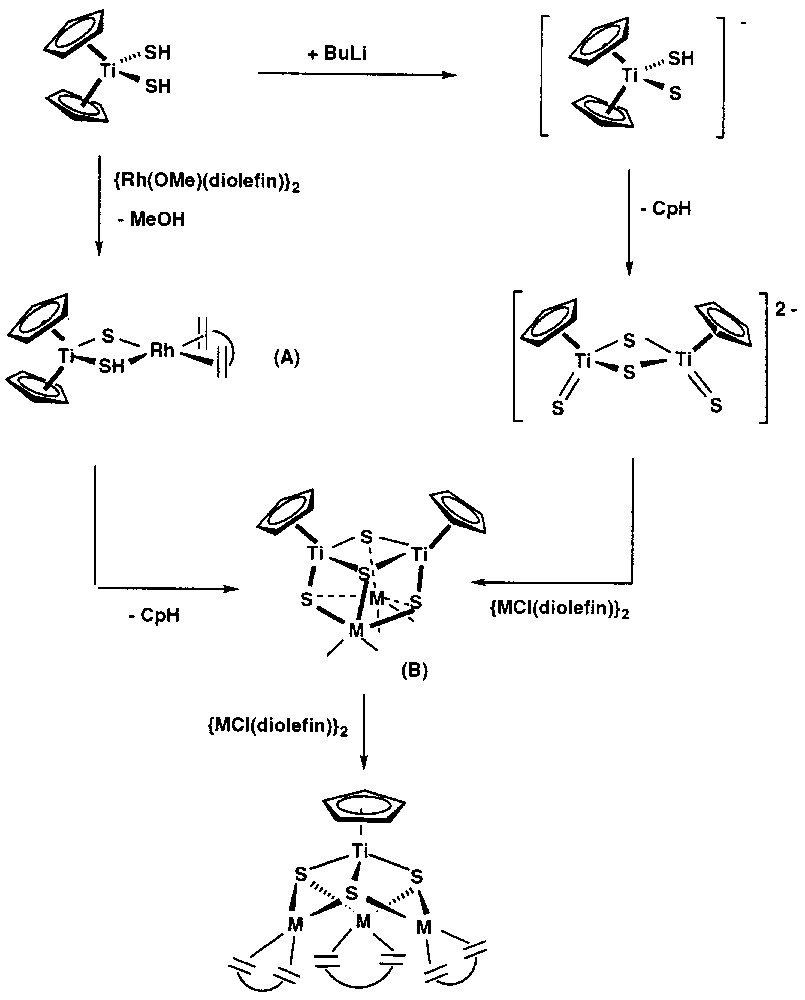
Reactions leading to [CpTi(μ3-S)3{Rh(diolefin)}3] complexes from [Cp2Ti(SH)2] with the methoxorhodium complexes.
2.2 Heterobimetallic TiRh3 and TiIr3 carbonyl complexes
Replacement of the diolefin ligands in [CpTi(μ3-S)3{M(diolefin)}3] (M = Rh, Ir) complexes by carbon monoxide takes place to give the hexacarbonyl compounds [CpTi(μ3-S)3{M(CO)2}3] (M = Rh, Ir). The nuclearity and the incomplete cubane framework of the starting materials are maintained in these reactions [23–24]. Reactions of the hexacarbonyl iridium complex with a variety of phosphine and phosphite ligands occurred readily under evolution of carbon monoxide to give the complexes [CpTi(μ3-S)3Ir3(μ-CO)(CO)3(PR3)3] (PR3= PPh3, PMe3, P(OMe)3, PMePh2), containing a bridging carbonyl group, as evidenced by a ν(CO) band in the range 1770–1740 cm–1 in the IR spectra. Remarkable features in the structure of these compounds, as found for [CpTi(μ3-S)3Ir3(μ-CO)(CO)3{P(OMe3)3}] by X-ray diffraction studies, are a distorted tetrahedral metal framework with short iridium–iridium distances and a tetrahedral coordination of the iridium atom closest to the titanium. The tetrahedral coordination of one iridium atom, together with the equatorial disposition of the phosphite ligands and consequent axial disposition for the carbonyl ligands, and the metal–metal bonding in the triiridium triangle are crucial in explaining the dynamic and chemical behaviour observed for these complexes [23]. While the bridging carbonyl group moves along the triangle, all the iridium centres are successively exchanging their stereochemistry, becoming tetrahedral and interacting with the titanium centre. Moreover, the carbonyl exchange in these clusters with external CO is very fast, as deduced from 13CO-labeling NMR experiments. However, when the electron-rich tetrahedral iridium centre is protonated (Fig. 3), the carbonyl scrambling ceases [23].
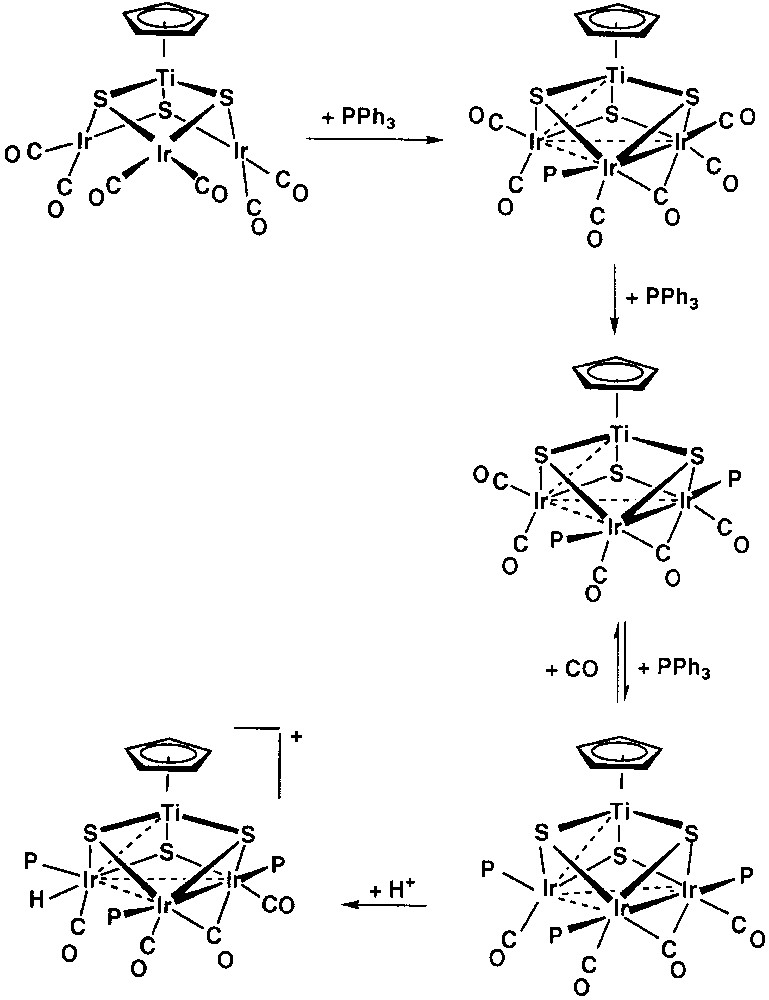
Reactions of the hexacarbonyl iridium complex with a variety of phosphine and phosphite ligands under carbon monoxide evolution, giving the complexes [CpTi(μ3-S)3Ir3(μ-CO)(CO)3(PR3)3] (PR3= PPh3, PMe3, P(OMe)3, PMePh2).
In contrast to the mentioned [CpTi(μ3-S)3Ir3(μ-CO)(CO)3(PR3)3] compounds, where a bridging carbonyl was present, a complex of formula [CpTi(μ3-S)3Ir3(CO)3(PMe2Ph)3], lacking the bridging carbonyl ligand, was isolated when PMe2Ph was used [23]. This type of complex is analogous to those resulting from the reactions of monodentate P-donor ligands with the rhodium complex [CpTi(μ3-S)3{Rh(CO)2}3]. These replacement reactions are highly selective. Thus, the more symmetrical C3 isomers of [CpTi(μ3-S)3{Rh(CO)(PR3)}3] are the only isolated product in the case of monodentate phosphine (PPh3, PMe3 or PCy3) and phosphite ligands (P(OPh)3, P(OMe)3 or P(o-OC6H4tBu)3) [31]. Nevertheless, detailed multinuclear NMR studies at low temperature have suggested that the formation of [CpTi(μ3-S)3{Rh(CO)(PPh3)}3] from [CpTi(μ3-S)3{Rh(CO)2}3] implied the intermediacy of 62-electron clusters, similar to the isolated iridium complexes, containing a bridging carbonyl ligand and a single Rh-Rh bond, as shown in Fig. 4.
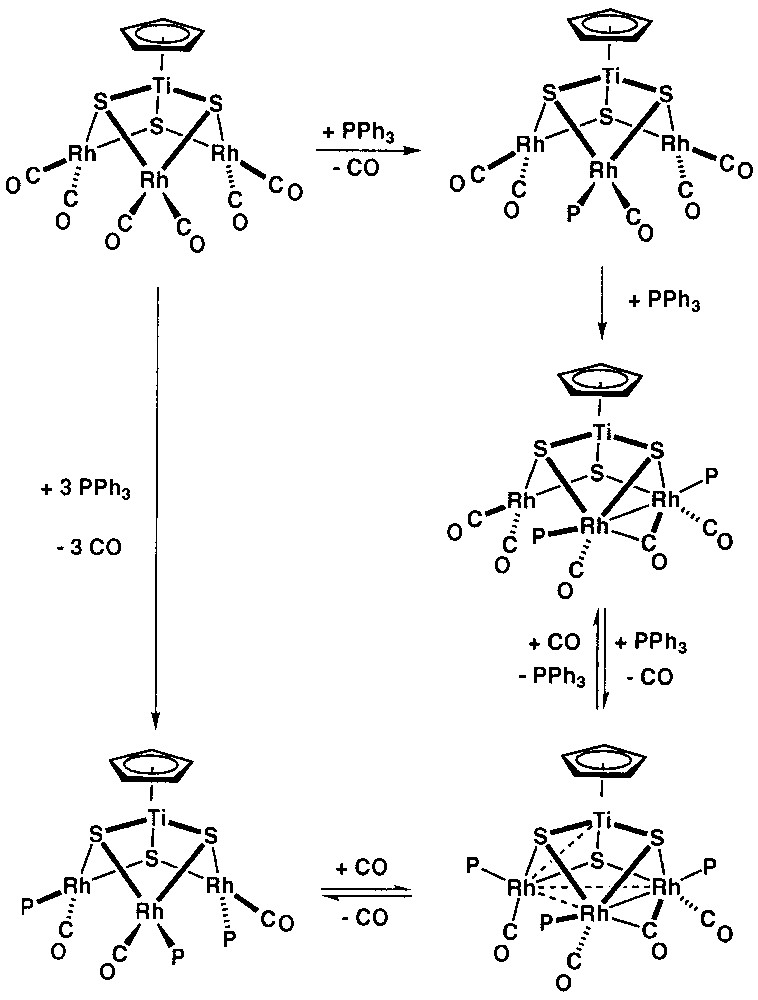
Formation of [CpTi(μ3-S)3{Rh(CO)(PPh3)}3] from [CpTi(μ3-S)3{Rh(CO)2}3] by the intermediacy of 62-electron clusters, similar to the isolated iridium complexes, containing a bridging carbonyl ligand and a single Rh-Rh bond.
The substitution reactions on [CpTi(μ3-S)3{Rh(CO)2}3] with diphosphines are strongly dependent on the nature of the diphosphine. Usually, only one molar equivalent of diphosphine was easily incorporated into the heterotetranuclear framework (dppp or BINAP); nevertheless, in some cases, the replacement of four of the carbonyl ligands allowed the introduction of two molar equivalents of 1,2-bis(diphenylphosphino)ethane (dppe). Thus, the reaction with one molar equivalent of dppe gave the complex [CpTi(μ3-S)3Rh3(CO)4(μ-dppe)], whilst reaction with two molar equivalents of the diphosphine gave [CpTi(μ3-S)3Rh3(μ-dppe)(CO)2(η2-dppe)] [31]. In the above-mentioned reactions, the metal ‘CpTi(μ3-S)3Rh3’ framework was maintained in all replacement transformations.
2.3 Heterobimetallic TiRh2 and TiIr2 diolefin complexes
We have commented above on the ability of [Cp2Ti(SH)2] to protonate appropriate d8 rhodium and iridium compounds yielding heterotetranuclear [CpTi(μ3-S)3{M(diolefin)}3] complexes, in which the group [CpTi(S)3]3– capped a trimetallic triangle of the late metals. However, if the late-metal complexes contain an anionic and good chelating ligand, this is transferred to the titanium, which results in a lower nuclearity of the early-late heteronuclear complexes. Thus, the reaction of [Cp2Ti(SH)2] with [M(acac)(diolefin)] (acac = acetylacetonate) yielded the heterotrinuclear complexes [Cp(acac)Ti(μ3-S)2{M(diolefin)}2] (M = Rh, diolefin = cod, tfbb; M = Ir, diolefin = cod) [32]. The formation of these heterobimetallic complexes resulted from a complex reaction involving the deprotonation of the hydrosulphido ligands along with the addition of the late-metal fragments, followed by the release of cyclopentadiene and the coordination of acetylacetonate to the titanium centre. Similarly, the reaction of [Cp2Ti(SH)2] with [M(quinol)(diolefin)] (quinol = 8-oxyquinolinate) afforded the complexes [Cp(quinol)Ti(μ3-S)2{M(diolefin)}2] (M = Rh, diolefin = cod, tfbb; M= Ir, diolefin = cod), which incorporate the 8-oxyquinolinate ligand (Fig. 5). These d0–d8 trinuclear early-late heterobimetallic complexes exhibit triangular TiRh2 and TiIr2 cores doubly capped by two μ3-sulphido ligands, as substantiated for [Cp(acac)Ti(μ3-S)2{M(diolefin)}2] by X-ray diffraction methods [32]. A tentative mechanism for the formation of the latter complex is presented in Fig. 6, being the first step to the monodeprotonation of [Cp2Ti(SH)2] by one equivalent of [M(acac)(diolefin)] to give dinuclear [Cp2Ti(μ-S)(μ-SH)M(diolefin)] species, followed by a further reaction with another equivalent of [M(acac)(diolefin)]. However, whether the [Cp2Ti(μ-S)2{M(diolefin)}2] complexes are initially formed and then they react with acacH or the release of CpH takes place before the addition of the second late-metal centre is still unclear.
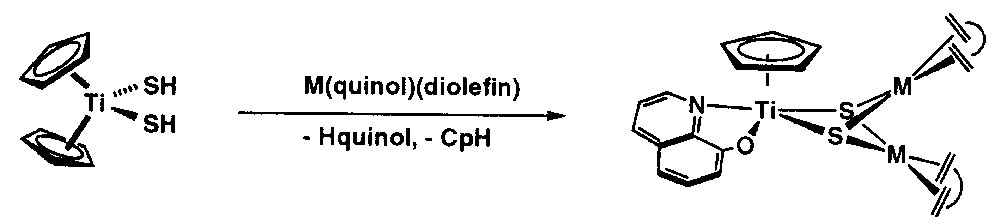
Reaction of [Cp2Ti(SH)2] with [M(acac)(diolefin)] (acac = acetylacetonate) yielding the heterotrinuclear complexes [Cp(acac)Ti(μ3-S)2{M(diolefin)}2] (M = Rh, diolefin = cod, tfbb; M= Ir, diolefin = cod).
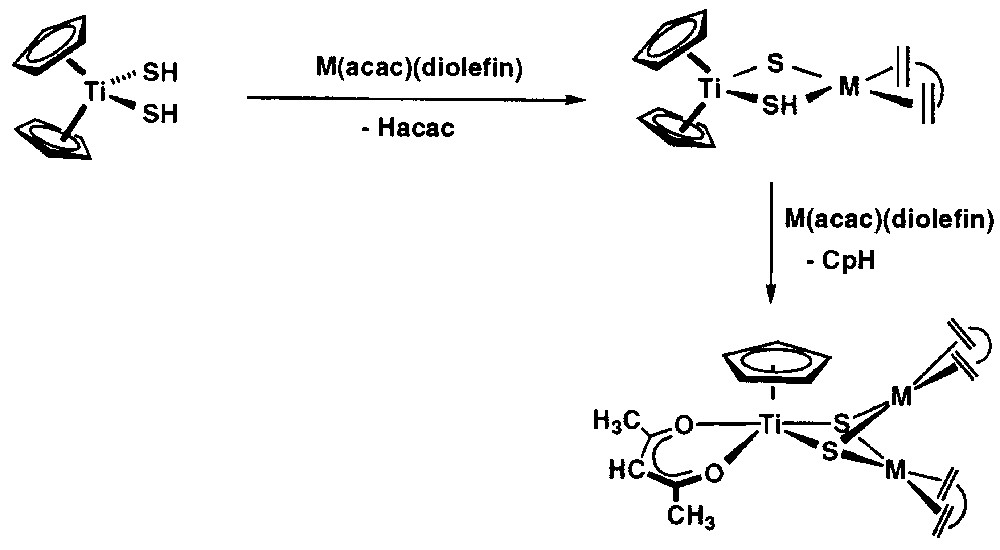
Monodeprotonation of [Cp2Ti(SH)2] by one equivalent of [M(acac)(diolefin)] giving dinuclear [Cp2Ti(μ-S)(μ-SH)M(diolefin)] species, followed by a further reaction with another equivalent of [M(acac)(diolefin)].
2.4 Heterobimetallic TiRh2 and TiIr2 carbonyl complexes
The heterobimetallic complexes [Cp(acac)Ti(μ3-S)2{M(cod)}2] (M = Rh, Ir) reacted with carbon monoxide under atmospheric pressure, in dichloromethane, to give the [Cp(acac)Ti(μ3-S)2{M(CO)2}2] complexes. Similarly, the related 8-oxyquinolinate heterotrinuclear complexes [Cp(quinol)Ti(μ3-S)2{M(CO)2}2] were obtained by carbonylation of the diolefin [Cp(quinol)Ti(μ3-S)2{M(cod)}2] complexes.
The reaction of [Cp(acac)Ti(μ3-S)2{M(CO)2}2] with two molar equivalents of PPh3 afforded the complex [Cp(acac)Ti(μ3-S)2{M(CO)(PPh3)}2] after evolution of carbon monoxide (Fig. 7). The rhodium complex was obtained as a 3:5 mixture of the cis and trans isomers, while the formation of a 1:1 mixture was observed for the iridium derivative. The related rhodium and iridium 8-oxyquinolinate complexes [Cp(quinol)Ti((μ3-S)2{M(CO)(PPh3)}2] were conveniently prepared by reaction of the corresponding tetracarbonyl derivatives [Cp(quinol)Ti((μ3-S)2{M(CO)2}2] with two molar equivalents of PPh3. As observed for the acetylacetonate compounds, the formation of both complexes was not selective, since the expected four isomers arising from the mutual disposition of the PPh3 ligands as well as their relative disposition to the 8-oxyquinolinate ligand were observed.
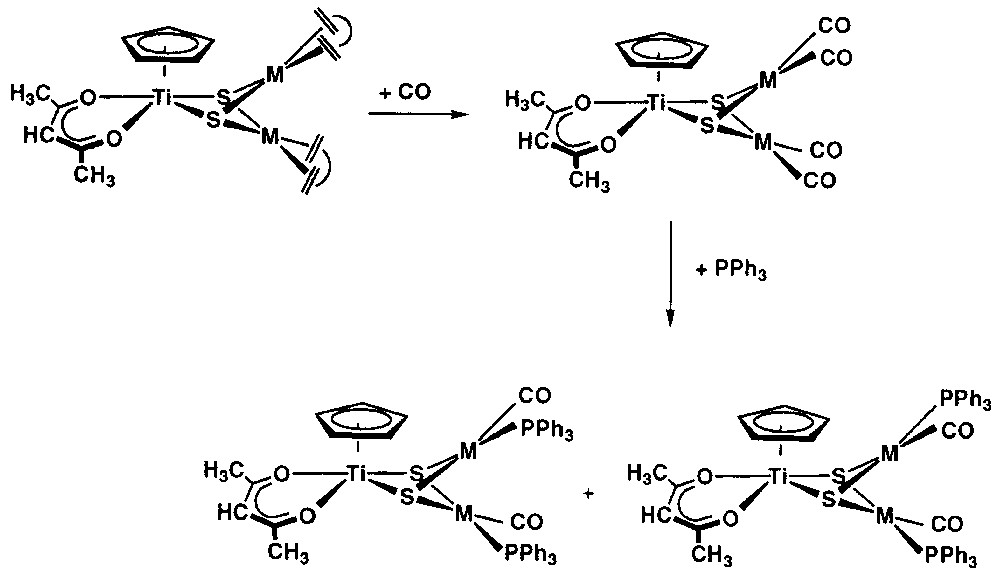
Reaction of [Cp(acac)Ti(μ3-S)2{M(CO)2}2] with two molar equivalents of PPh3affording the complex [Cp(acac)Ti(μ3-S)2{M(CO)(PPh3)}2] after evolution of carbon monoxide.
While the reaction of [Cp2Ti(SH)2] with [M(acac)(diolefin)] gave [Cp(acac)Ti((μ3-S)2{M(diolefin)}2], the analogous reaction with [M(acac)(CO)2] (1:3 molar ratio) in dichloromethane gave the ion-pair product [Cp2Ti(acac)][M3(μ3-S)2(CO)6]. In particular, the rhodium complex [Cp2Ti(acac)][Rh3(μ3-S)2(CO)6] was characterised by a X-ray diffraction study. These ion-pair complexes were most probably formed by the intermediacy of an unstable [Cp2Ti(μ3-S)2{M(CO)2}2] species, retaining two Cp ligands coordinated to titanium, which upon reaction with [M(acac)(CO)2] undergoes a sulphido ligand transfer according to Fig. 8. The anions [M3(μ3-S)2(CO)6]– (M = Rh and Ir) have been previously reported and structurally characterised [33].
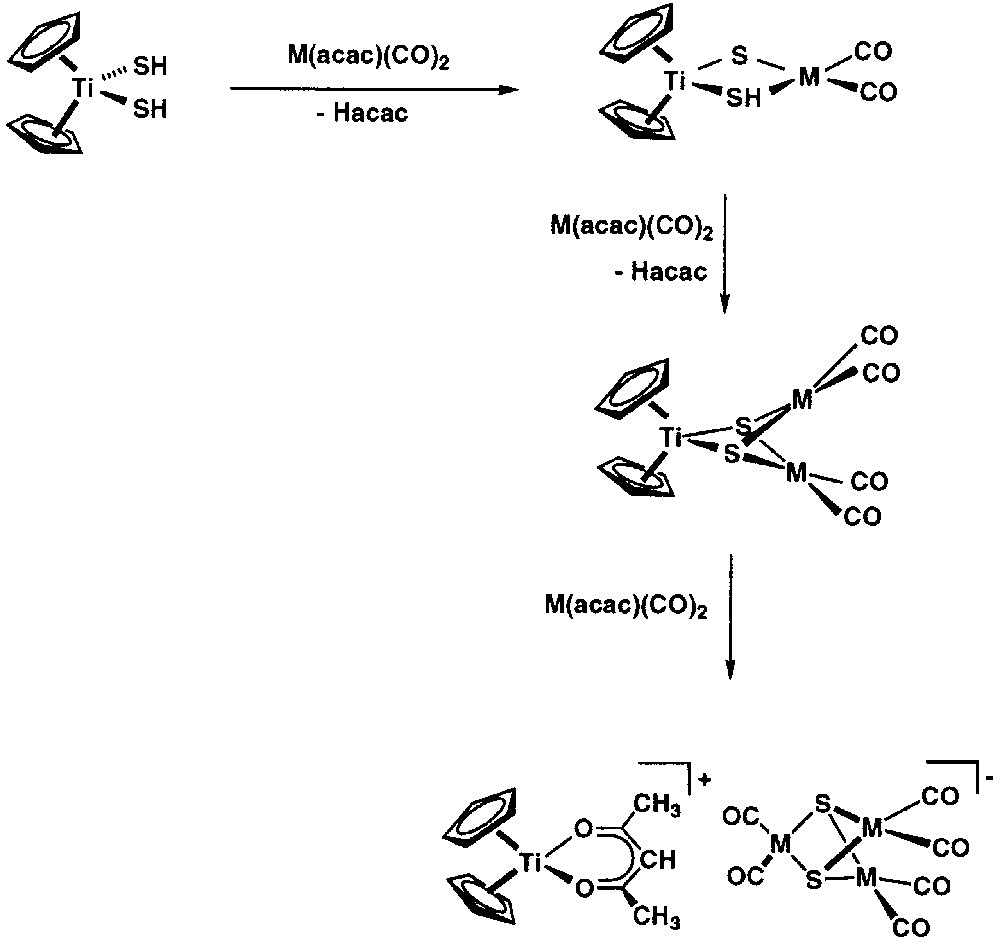
Formation of ion-pair complexes by the intermediacy of an unstable [Cp2Ti(μ3-S)2{M(CO)2}2] species, retaining two Cp ligands coordinated to titanium, which upon reaction with [M(acac)(CO)2] undergoes a sulphido ligand transfer.
2.5 Heterobimetallic Ti2Rh4 carbonyl complexes
The above-mentioned [CpTi(μ3-S)3{Rh(CO)2}3] complex presenting an incomplete cubane framework can easily be prepared by carbonylation of [CpTi(μ3-S)3{Rh(cod)}3], prepared in situ by addition of [{Rh(μ-OMe)(diolefin)}2] to [Cp2Ti(SH)2] (Fig. 9). However, when the reaction was carried out with either not carefully dried [{Rh(μ-OMe)(diolefin)}2], or in the presence of hydrated sodium carbonate, a novel oxo sulphido heteronuclear cluster of formula [(CpTi)2(μ4-O)(μ3-S)4{Rh4(CO)6}] was obtained. This complex reacted with a variety of P-donor ligands to give complexes of formula [(CpTi)2(μ4-O)(μ3-S)4{Rh4(CO)4(PR3)2}] (PR3 = PPh3, P(OMe)3, P(OPh)3). These unusual oxo sulphido complexes showed an incomplete double cubane-type structure formed by two cubane-type moieties ‘Rh2TiS2O’, each with one empty vertex-fused through a common ‘Rh2O’ face, with the oxide ligand displaying an unusual tetracoordination, as confirmed by X-ray diffraction methods for the triphenylphosphite derivative (Fig. 9) [34]. Most probably, the oxide ligand, bridging the two titanium centres, should be formed by reaction with the water present in the reaction medium. The oxophilic character of titanium and the presence of water played a key role in the formation of the oxo cluster. The replacement of the carbonyl groups in [(CpTi)2(μ4-O)(μ3-S)4{Rh4(CO)6}] by P-donor ligands occurred selectively at the two rhodium atoms possessing two sulphido ligands in a cis disposition. Very recently, a Ti2Ru2Pd2 hexanuclear oxo sulphido cluster of formula [(CpTi)2(Cp*Ru)2Pd2(PPh3)(μ3-S)4(μ3-O)(μ2-H)2] has been prepared by reacting the heterotrinuclear cluster [(CpTiCl2(Cp*Ru){Pd2(PPh3)2}(μ3-S)(μ2-S)(μ2-H)] with water [35].
Synthesis of the [CpTi(μ3-S)3{Rh(CO)2}3] complex presenting an incomplete cubane framework by carbonylation of [CpTi(μ3-S)3{Rh(cod)}3], prepared in situ by addition of [{Rh(μ-OMe)(diolefin)}2] to [Cp2Ti(SH)2].
3 Sulphido-bridged zirconium–rhodium and zirconium–iridium complexes
3.1 Heterobimetallic ZrRh2 and ZrIr2 diolefin complexes
We have previously commented that the [Cp2Ti(SH)2] complex is an appropriate starting material for the synthesis of early-late complexes of formula [CpTi(μ3-S)3{M(diolefin)}3]. Fig. 2 shows that the loss of one Cp ring is commonly observed. Concerning zirconium, some initial experiments with the related hydrosulphido complex [Cp2Zr(SH)2] resulted in extensive sulphido transfer to the d8 metal centres, giving the trimetallic complexes [M3(μ-S)2(μ-H)(cod)3] or the anionic clusters [M3(μ3-S)2(CO)6]– (M = Rh, Ir) as the main outcome of these reactions. We thought that the known dimerisation [36] of [Cp2Zr(SH)2] with extrusion of H2S probably complicated these reactions. To overcome this, we envisaged that the use of a bulkier zirconium-metallocene hydrosulphido compound, such as [Cptt2Zr(SH)2] (Cptt = η5-1,3-di-tert-butylcyclopentadienyl), could be useful to avoid the undesirable reactions and facilitate the controlled construction of new heterobimetallic ZrRh2 and ZrIr2 complexes. The [Cptt2Zr(SH)2] complex shows a remarkable stability with respect to the elimination of H2S when compared with the analogous compound [Cp2Zr(SH)2], probably because of the steric shielding of the metal atom given by the bulky substituents on the cyclopentadienyl rings.
The bishydrosulphido complex [Cptt2Zr(SH)2] was prepared by reacting [Cptt2ZrMe2], obtained by treatment of [Cptt2ZrCl2] with MeLi, with H2S under pressure [37]. Additive deprotonation of [Cptt2Zr(SH)2] with appropriate rhodium complexes, such as [Rh(acac)(diolefin)], results in the clean formation of heterotrinuclear complexes of formula [Cptt2Zr(μ3-S)2{Rh(diolefin)}2], without the loss of any coordinated cyclopentadienyl ligand (Fig. 10), although the cycloctadiene derivative that resulted was contaminated with [Rh3(μ-S)2(μ-H)(cod)3]. Nevertheless, the [Cptt2Zr(μ3-S)2{Rh(cod)}2] compound can be effectively prepared by reacting [Cptt2Zr(SH)2] with [{Rh(μ-OH)(cod)}2]. On the other hand, the reaction of [Cptt2Zr(SH)2] with two molar equivalents of [Ir(acac)(cod)] afforded the new complex [Cptt(acac)Zr(μ3-S)2{Ir(cod)}2] contaminated with [Ir3(μ-S)2(μ-H)(cod)3], which could be isolated pure by recrystallisation. This reaction implies the replacement of one cyclopentadienyl ring by the acetylacetonate ligand in the zirconium coordination sphere [38]. Related TiRh2 complexes have been described in section 2.3.
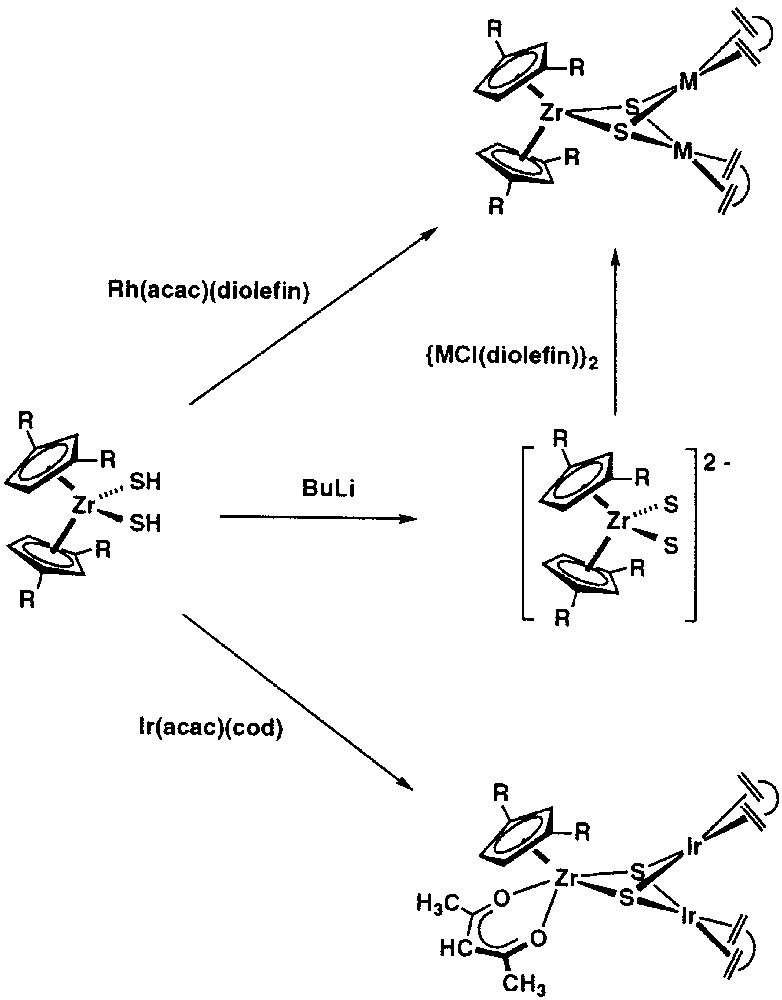
Additive deprotonation of [Cptt2Zr(SH)2] with appropriate rhodium complexes, such as [Rh(acac)(diolefin)], resulting in the clean formation of heterotrinuclear complexes of formula [Cptt2Zr(μ3-S)2{Rh(diolefin)}2], without the loss of any coordinated cyclopentadienyl ligand.
A general method for the synthesis of [Cptt2Zr(μ3-S)2{M(diolefin)}2] complexes (M = Rh, diolefin = nbd, cod; M = Ir, diolefin = cod) consisted in the reaction of [{M(μ-Cl)(diolefin)}2] compounds with the zirconium-sulphide metallocene anion [Cptt2ZrS2]2-, generated by double deprotonation of [Cptt2Zr(SH)2] with BuLi (Fig. 10). These compounds exist in solution as two rotamers, eclipsed and staggered, due to a hindered rotation of the cyclopentadienyl rings and the relative disposition of the Cptt groups in the sandwich moiety [38]. The two rotamers interconvert for the complexes with diolefin = nbd and tfbb, whilst no interconversion occurs for the cod complexes, which can be isolated as the pure staggered rotamer.
3.2 Heterobimetallic ZrRh2 and ZrIr2 carbonyl complexes
Carbonylation of [Cptt2Zr(μ3-S)2{Rh(nbd)}2] under atmospheric pressure gave the carbonyl complex [Cptt2Zr(μ3-S)2{Rh(CO)2}2]. This complex was also prepared by reacting [Cptt2Zr(SH)2] with a variety of rhodium–carbonyl complexes. Thus, the reactions of [Cptt2Zr(SH)2] with two molar equivalents of [Rh(acac)(CO)2], or with [{Rh(μ-Cl)(CO)2}2] in the presence of a slight excess of NEt3, gave [Cptt2Zr(μ3-S)2{Rh(CO)2}2]. The ZrRh2core of the compound [Cptt2Zr(μ3-S)2{Rh(CO)2}2] has been established by X-ray diffraction methods [37–38]. The related iridium carbonyl complex [Cptt2Zr(μ3-S)2{Ir(CO)2}2] was obtained from the reaction of [Cptt2Zr(SH)2] with two molar equivalents of the anionic complex [IrCl2(CO)2]– in the presence of a slight excess of NEt3 (Fig. 11 ) [39]. This [Cptt2Zr(μ3-S)2{Ir(CO)2}2] complex reacted with 1,2-bis(diphenylphosphino)ethane (dppe) yielding selectively the ion-pair [Ir(CO)(dppe)2][Cptt2Zr(μ-S)2Ir(CO)2]. A related heterobimetallic anion [Cp*2Zr(μ-S)2Rh(CO)2]– has been previously reported [40]. Apart from the above-mentioned reaction, the ZrM2 triangular core in the heterotrinuclear carbonyl clusters [Cptt2Zr(μ3-S)2{M(CO)2}2] (M = Rh, Ir) is usually maintained in the replacement reactions of carbonyl groups by P-donor ligands. The outcome of these reactions depends on the character of the P-donor ligands. Thus, the replacement reactions with the short-bite bidentate bis(diphenylphosphino)methane (dppm) ligand are stereoselective, and the products [Cptt2Zr(μ3-S)2{M(CO)}2(μ-dppm)] are cleanly obtained as a single isomer. In contrast, the disubstituted complexes [Cptt2Zr(μ3-S)2{Rh(CO)(P(OR)3)}2] (P(OR)3 = P(OMe)3 and P(OPh)3) are obtained as mixtures of the transoid and cisoid isomers, which exhibit a restricted rotation of the Cptt rings [38].
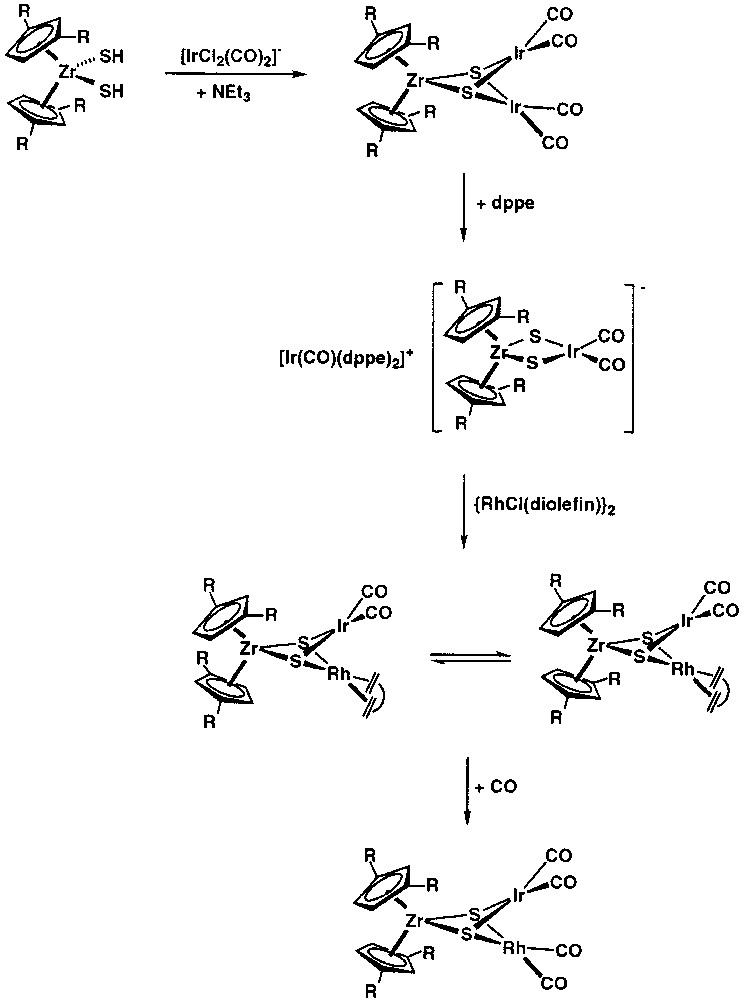
Synthesis of the iridium carbonyl complex [Cptt2Zr(μ3-S)2{Ir(CO)2}2] by the reaction of [Cptt2Zr(SH)2] with two molar equivalents of the anionic complex [IrCl2(CO)2]–, in the presence of a slight excess of NEt3.
3.3 Heterotrimetallic ZrIrRh carbonyl complexes
Interestingly, the heterodinuclear anion [Cptt2Zr(μ-S)2Ir(CO)2]– is capable of acting as a metalloligand towards d8 metal centres allowing the syntheses of novel Zr–Ir–Rh heterotrimetallic complexes (Fig. 11). Thus, this anion reacted with [{Rh(μ-Cl)(cod)}2], yielding the complex [Cptt2Zr(μ3-S)2{Ir(CO)2}{Rh(cod)}], which has been characterised by X-ray diffraction methods. The structure showed a triangular ZrIrRh core capped on both sides by two symmetrical μ3-sulphido ligands. Although no significant d0–d8 bonding interactions are detected, the Rh···Ir distance, 2.8205(10) Å, suggested the presence of a significative d8–d8 metal–metal interaction. A remarkable feature of this heterometallic complex is the fully staggered relative disposition of the Cptt ligands observed in the solid state, while in CDCl3 solution two interconverting rotamers are detected in a 3:1 ratio. The major rotamer showed a staggered disposition of the tert -butyl groups, as found in the solid state, whilst the spectroscopic information for the minor isomer suggested an eclipsed conformation. As expected, the carbonylation of [Cptt2Zr(μ3-S)2{Ir(CO)2}{Rh(cod)}] gave the heterotrimetallic compound [Cptt2Zr(μ3-S)2{Ir(CO)2}{Rh(CO)2}] (Fig. 11). This complex can also be obtained in one-pot synthesis by successive additions of dppe and the anion [RhCl2(CO)2]– to the heterobimetallic [Cptt2Zr(μ3-S)2{Ir(CO)2}2] compound [39].
4 Catalytic activity
The importance of heterometallic complexes for homogeneous catalysis is based on the reasonable expectation that two or more adjacent metal centres may offer possibilities for cooperative reactivity and synergism. In particular, some early-late Zr–Rh and Ti–Rh heterobimetallic systems have been shown to be efficient hydroformylation catalysts [41–43].
The heterotetranuclear [CpTi(μ3-S)3{Rh(diolefin)}3] complexes, in the presence of phosphorus ligands, were found to be active catalyst precursors in the hydroformylation of 1-hexene and styrene. Thus, 1-hexene was hydroformylated at 5 bar and 353 K (P/Rh ratio = 2 or 4) with a 96% conversion to aldehydes and a 77% regioselectivity for the linear aldehyde. A higher P/Rh ratio provided a better selectivity for the linear aldehyde (85%), although lower conversions were obtained. Taking into account the equilibrium observed between the different heterotetranuclear species containing carbonyl and phosphine ligands (Fig. 4), and in order to know which of those intermediates were formed under CO pressure, a high-pressure NMR-spectroscopy study was carried out. Thus, the 31P{1H} NMR spectrum of a solution of [CpTi(μ3-S)3{Rh(cod)}3] and PPh3(P/Rh = 2), after being pressurised to 2.5 bar of carbon monoxide, showed an equilibrium between the species [CpTi(μ3-S)3Rh3(μ-CO)(CO)4(PPh3)2] and [CpTi(μ3-S)3Rh3(μ-CO)(CO)3(PPh3)3] at 213 K. The latter species became predominant when the P/Rh ratio was increased to 6. Most probably, the variation of the active species could be the reason for the higher selectivity and lower conversion observed using this P/Rh ratio in the hydroformylation reaction. Interestingly, the 31P{1H} NMR and IR spectra of the solutions obtained at the end of the hydroformylation experiments showed the presence of the heterotetranuclear [CpTi(μ3-S)3{Rh(CO)(PPh3)}3] species, indicating that the heterotetranuclear framework is maintained under hydroformylation conditions.
The complex [Cptt2Zr(μ3-S)2{Rh(CO)2}2] in the presence of monodentate P-donor ligands was also an active catalyst precursor for hydroformylation of oct-1-ene under mild conditions of pressure and temperature. The catalytic systems obtained with phosphite ligands were much more active than the corresponding with triphenylphosphine. In addition, the system formed with trimethylphosphite was much more selective than the system obtained with triphenylphosphite, giving an aldehyde selectivity of 95% with a regioslectivity close to 80%. The spectroscopic analysis of the solutions obtained after the hydroformylation reactions with the [Cptt2Zr(μ3-S)2{Rh(CO)2}2] precursor showed no evidence for the presence of the complexes [Cptt2Zr(μ3-S)2{Rh(CO)(P(OR)3)}2], indicating that the heterotrinuclear complexes are not maintained as such, and most probably break down to active monomeric rhodium species. This behaviour is opposite to that observed in the hydroformylation of alkenes using the catalyst precursor [CpTi(μ3-S)3{Rh(diolefin)}3], indicating an outstanding stability of the heterotetranuclear framework.
5 Concluding remarks
The controlled syntheses of novel d0–d8early-late tri-, tetra-, and hexa-nuclear diolefin and carbonyl clusters with the cores TiRh2, TiIr2, ZrRh2, ZrIr2, ZrRhIr, TiRh3, TiIr3, and Ti2Rh4, were accomplished through additive-deprotonation reactions involving the titanium and zirconium bis-hydrosulphido complexes of formula [Cp2Ti(SH)2] and [Cptt2Zr(SH)2] and appropriate rhodium and iridium diolefin and carbonyl compounds. Some of the titanium–rhodium and zirconium–rhodium complexes are active catalyst precursors for hydroformylation of alkenes. All the observations pointed out that the heterotetranuclear ‘CpTi(μ3-S)3Rh3’ framework was maintained under hydroformylation conditions.