

1 Introduction
The intramolecular addition of a zinc ester enolate onto an unactivated double bond has recently been reported simultaneously in our [1–5] and other laboratories [6–9]. This unusual reaction concerns the reaction of a Reformatsky-type reagent 1 on a terminal double bond leading to an alkyzinc reagent 2, which then can be further functionalised to 3 through reaction with electrophiles (Fig. 1). This reaction is interesting both on the mechanistic and synthetic points of view. On the one hand, this reaction apparently seems contra-thermodynamic, since leading to an alkylmetal reagent from a starting material (enolate) with a lower basicity. On the other hand, this reaction allows the preparation of piperidines and pyrrolidines with an excellent diastereomeric and enantiomeric control, and then a straightforward route to substituted α- and β-prolines and pipecolic acids.
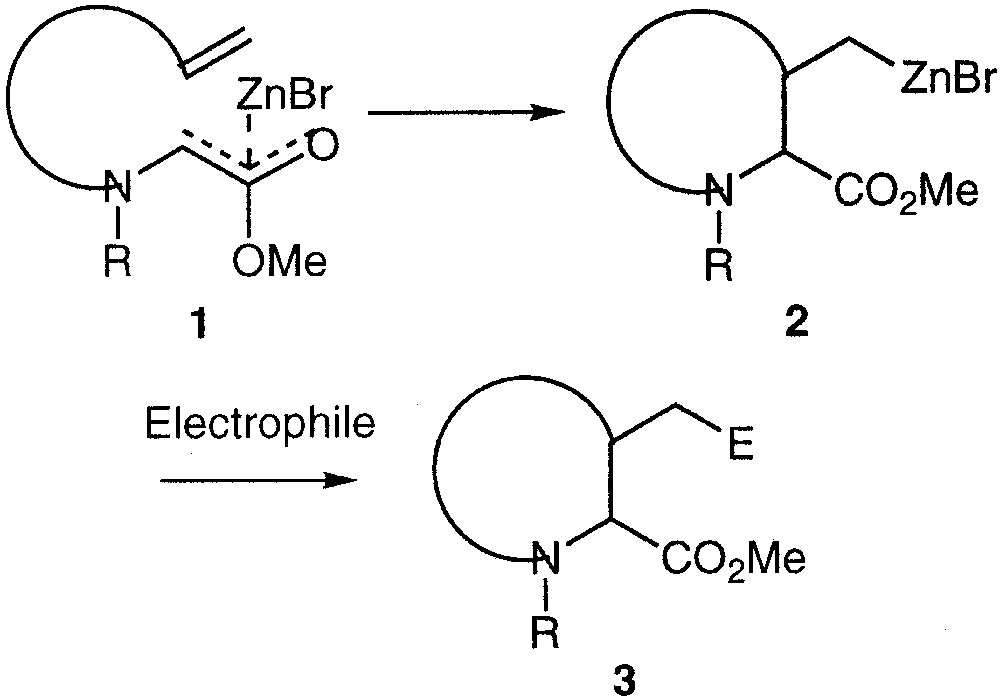
Carbocyclisation reactions of zinc enolates.
Although inter- and intramolecular carbometalation reactions (addition of organometallic species onto unactivated multiple bonds) have been widely developed [10,11], much little is known concerning the carbometalation of unactivated alkenes by stabilized enolate-type organometallics. Addition of ketones and malonates on styrenes under t-BuOK catalysis [12] is reported, as well as the intramolecular cyclisation of lithium enolates on (η2-alkene)Fe complexes [13–16], (η4-diene)Fe complexes [17–19], and of sodium malonates on (η4-diene)Mo complexes [20–22]. Relatively few examples are involving totally unactivated alkenes: addition of enolates or malonates on alkenes in the presence of Pd(II) salts or under Pd(0) catalysis [23,24] have been described; in most cases, the organometallic species resulting from the carbometalation reaction undergoes dehydropalladation or reductive elimination. On the other hand, inter- or intramolecular carbometalations of unactivated olefins have been reported with tin malonates [25,26] and zincated hydrazones [27–30]. In these cases, the organometallic reagent resulting from the carbocyclisation process can be further functionalised by an external electrophile added at the end of the reaction.
We first present an overview of our previous results concerning the reaction of α-(N-homoallyl)-aminoesters leading to 2-carbomethoxy-pyrrolidines, of α-(N-4-pentenyl)-aminoesters leading to 2-carbomethoxy-piperidines and of β-(N-allyl)-aminoesters leading to 3-carbomethoxy-pyrrolidines, and then our latest results concerning the discrepancies of the steric outcome of these cyclisations.
2 Carbocyclisation reactions
2.1 Carbocyclisation reactions of α-(N-homoallyl)-aminoesters [1,3]
The deprotonation of α-(N-benzyl-N-homoallyl)-amino methylester 4 with LDA in ether followed by the addition of zinc bromide results in a smooth diastereoselective carbocyclisation leading to the cis-3-methyl-N-benzyl-2-carbomethoxy pyrrolidine 5 after hydrolysis (Fig. 2). The intermediacy of the 3-pyrrolidinylmethylzinc compound 6 in this reaction was shown by its reaction with various electrophiles. The diastereoselectivity of this carbocyclisation was attributed to a zinc-enolate-ene-type reactive intermediate 7, where the double bond of the O-centred enolate is eclipsing the terminal reacting double bond, giving thus the observed cis stereoselectivity.
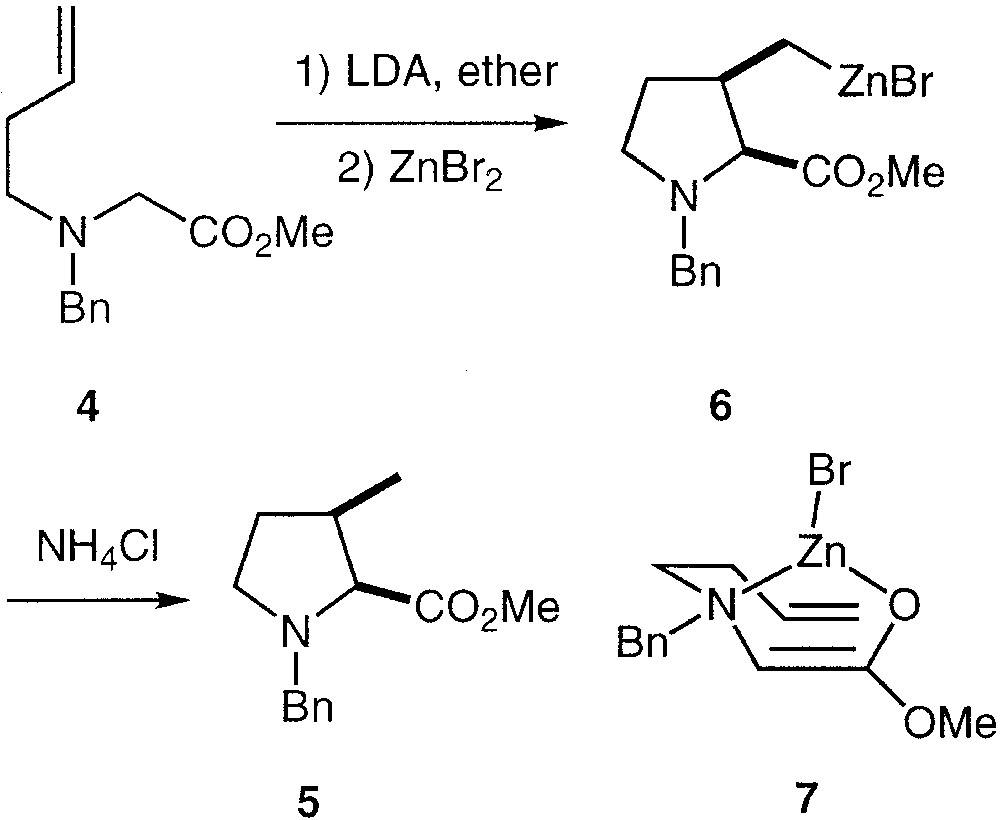
Diastereoselectivity in the carbocyclisation reaction.
The Z-configuration of the enolate is dictated by the Zn–N coordination, which has been observed [31] both in solution and in the solid state for analogous Reformatsky reagents derived from N,N-silicon disubstituted α-aminoesters.
The presence of a substituent on the homoallyl chain is directing the diastereoselectivity of the reaction, the reactive intermediate in which this substituent is occupying the pseudo equatorial position (for an alkyl group) or the pseudoaxial position (for an acetal group) leading to the major observed diastereomer (Fig. 3, reaction of aminoesters 8, 9 and 10). When two substituents are present, as for the aminoester 11, the homoallylic substituent was found to direct totally the diastereoselectivity.

Diastereoselectivity in the 2-carbomethoxypyrrolidine formation.
In all cases, the resulting pyrrolidine is presenting a cis relationship between the carbomethoxy group and the methyl group coming from the methylzinc bromide moiety. This stereochemical feature was again attributed to the O-centred zinc-enolate-ene-type reactive intermediate.
2.2 Carbocyclisation reactions of α-(N-4-pentenyl)-aminoesters [2]
The carbocyclisation reaction of zinc enolates derived from α-(N-4-pentenyl)-aminoesters as 12 is much slower than in the case of α-(N-homoallyl)-aminoesters, but the diastereoselectivity is excellent and the corresponding cis-2-carbomethoxy-3-methylpiperidine 13 is obtained as a single diastereomer. Here again, the organozinc reagent coming from the carbocyclisation reaction can be easily functionalised with various electrophiles. An oxygen-centred zinc-enolate-ene-type reactive intermediate as 14 (Fig. 4) was proposed, in order to explain the very high cis selectivity. The presence of a substituent in the homoallylic position (Fig. 4, aminoester 15) results in the stereoselective construction of a trisubstituted piperidine through the reactive intermediate presenting the substituent in an equatorial position. However, when a substituent is present in both allylic and homoallylic positions (Fig. 4, aminoesters 16 and 17), only the homoallylic one is directing the diastereoselectivity of the reaction. It is also directing the diastereoselectivity when a substituent is present in both homoallylic position and α to the nitrogen atom (Fig. 4, aminoester 18). Finally, when three substituents are present in the allylic, homoallylic and α to nitrogen positions, the relative stereochemistries of the two latter being chosen to counterbalance the one of the former (Fig. 4, aminoester 19), the resulting pentasubstituted piperidine was obtained with a low diastereoselectivity.

Diastereoselectivity in the 2-carbomethoxypiperidine formation.
2.3 Carbocyclisation reactions of β-(N-allyl)-aminoesters [4]
The carbocyclisation reaction of zinc enolates derived from β-(N-allyl)-aminoesters is more difficult than the reaction on α-(N-homoallyl)-aminoesters due to the competing β-elimination reaction. However, for β-(N-allyl)-aminoesters 21 bearing a substituent α to the ester group, we have recently shown that this side-reaction is limited and carbocyclisation occurs smoothly to give the corresponding 3-carbomethoxy-4-methylzinc pyrrolidine reagent 22 (Fig. 5), and (in good yield) the pyrrolidine 23 upon hydrolysis. Here again, the intermediacy of the 4-pyrrolidinylmethylzinc compounds 22 in this reaction was shown by their reactions with various electrophiles.

Carbocyclisation reaction of substituted β-(N-allyl)-aminoesters.
Surprisingly, the observed high diastereoselectivity was found in favour of the trans pyrrolidine. This diastereoselectivity cannot be explained by an O-centred zinc-enolate-ene-type reactive intermediate, as in the case of α-(N-homoallyl)-aminoesters, as the latter leads to cis pyrrolidines. We have proposed a C-centred zinc-enolate-carbometallation-type reactive intermediate 24 (Fig. 5) to explain the observed diastereoselectivity. The main feature of this reactive intermediate is the pseudoaxial position of the carbomethoxy group due to (i) simple steric considerations, (ii) possible chelation of an extra zinc salt by nitrogen and the sp2 oxygen of the carbomethoxy moiety.
The reaction of the parent unsubstituted β-(N-allyl)-aminoester 25 leads only to a β-elimination product. However, we have found that this unwanted reaction is limited by the use of zinc iodide for the Reformatsky preparation as well as by a ‘reverse-addition’ procedure of lithium enolate on zinc iodide. The diastereoselectivity is not as high as for 21, but is still good as the corresponding pyrrolidine 26 is obtained in an 87:13 ratio in favour of the trans diastereomer (Fig. 6). For the substituted case 27, a disappointing mixture of three diastereomers was obtained. By contrast, in the disubstituted case 28, a very good diastereoselectivity was observed. However, lower diastereoselectivities were obtained in the disubstituted cases 29 and 30. Finally, a mixture of three diastereomers in an 83:12:5 ratio was obtained starting from 31, but the relative stereochemistries of the chiral centres in the major diastereomer could not be ascertained.
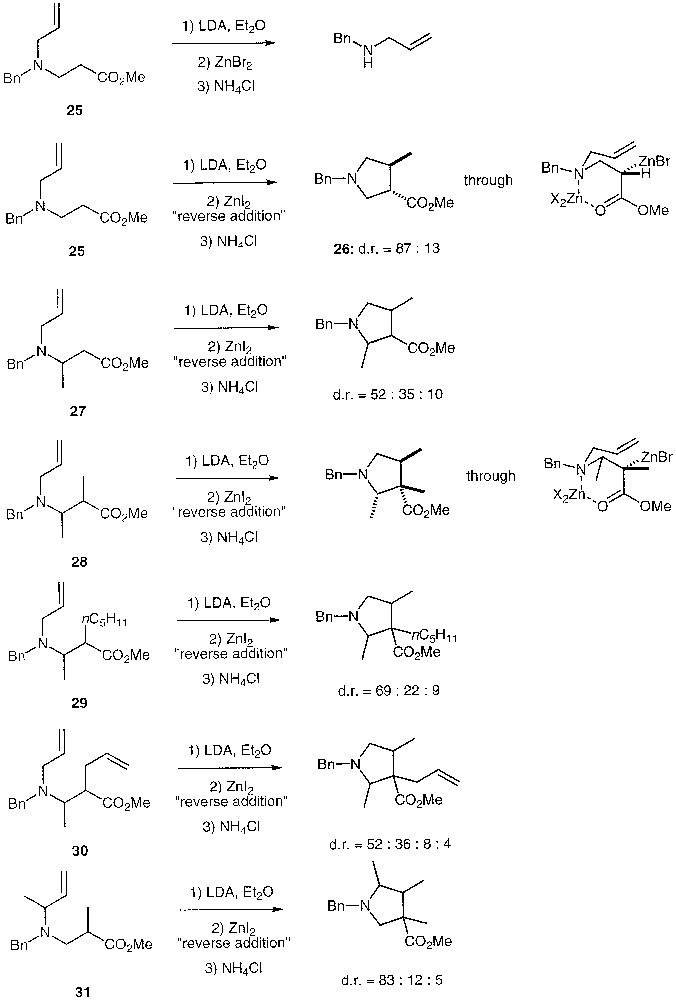
Carbocyclisation reactions of various β-(N-allyl)-aminoester.
2.4 Pyrrolidine formation by domino Michael addition/carbocyclisation reaction [5]
In the course of our studies toward the carbocyclisation reaction of β-(N-allyl)-aminoesters, we have shown that the above-described formation of 3,4-disubstituted-3-carbomethoxy pyrrolidines can be also obtained by the three-component one-pot domino reaction depicted in Fig. 7. 1,4-Addition of lithium zincate or higher-order cyanocuprate reagents on the common starting material 32 leads to the enolate 33. Further carbocyclisation reaction of 33 is observed upon the addition of zinc(II) salts to yield the metalated pyrrolidines 34, which lead upon hydrolysis to the diversely 3,4-disubstituted 3-carbomethoxypyrrolidines 35. This interesting domino reaction can also be performed in one step by using copper–zinc reagents. The resulting metalated pyrrolidine intermediate could be functionalised with various electrophiles to pyrrolidines 36.

Domino Michael addition/carbocyclisation reaction.
In all cases, pyrrolidines are obtained with a good control of the diastereoselectivity, again in favour of the trans disubstituted diastereomer, as depicted in Fig. 8 for pyrrolidines 35 and 36. The diasteroselectivity can be again explained by a C-centred zinc or copper–zinc–enolate-carbometallation-type reactive intermediate, as for the pyrrolidines obtained through deprotonation of the substituted β-(N-allyl)-aminoesters.

Reactive intermediates for domino reactions.
3 Considerations upon C-metalated versus O-metalated enolate-ene reactive intermediates
The puzzling dichotomy observed in the stereochemical behaviour of α-(N-homoallyl)-aminoesters and β-(N-allyl)-aminoesters is explained on the basis of O-metalated or C-metalated enolate-ene transitions states. Zinc enolates derived from α-aminoesters have been found to be O-metalated [31] but, on the other hand, Reformatsky reagents have been shown to be C-metalated by NMR and X-ray crystallographic studies [32–35]. Moreover, the stereochemical outcome from α-(N-homoallyl)-aminoesters can be explained as well as by the original O-metalated transitions state TS1 (Fig. 9), as on the basis of the C-metalated enolate-ene reactive intermediate TS2 analogous to the reactive intermediate TS3 proposed in the carbocyclisation of β-(N-allyl)-aminoesters.
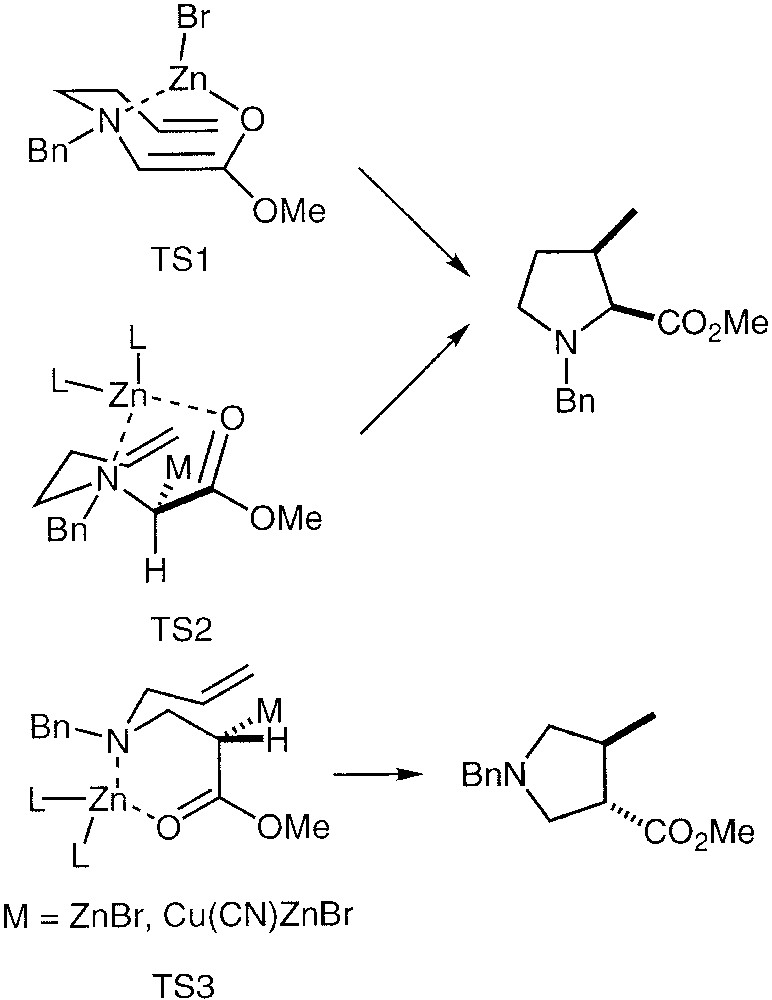
Possible C- and O-metalated reactive intermediates.
The choice between O- and C-metalated reactive intermediates leads to a concept far beyond this simple difference. The intrinsic mechanism of the carbometalation reaction is concerned, since the O-metalated reactive intermediate mainly looks like an enolate-ene six-membered reactive intermediate, whereas in the case of a C-metalated reactive intermediate, a four-membered reactive intermediate is presumably involved.
In order to know whether carbocyclisation reactions from α-(N-homoallyl)-aminoesters are involving C- or O-metalated reactive intermediate, we have examined the feasibility and the stereochemical course of carbocyclisation from aminoesters 38 (Fig. 10) [36]. If an O-metalated enolate is necessary for a carbocyclisation, TS4 should be favoured over TS6 on the basis of steric hindrance between the R residue on the nitrogen atom and the vinylic moiety. The trans metalated pyrrolidine 39 should then be obtained. By contrast, if a C-metalated enolate is involved, TS7 should be favoured over TS5 (because of the interaction between this R residue and the alkyl chain) to yield the cis-metalated pyrrolidine 40.
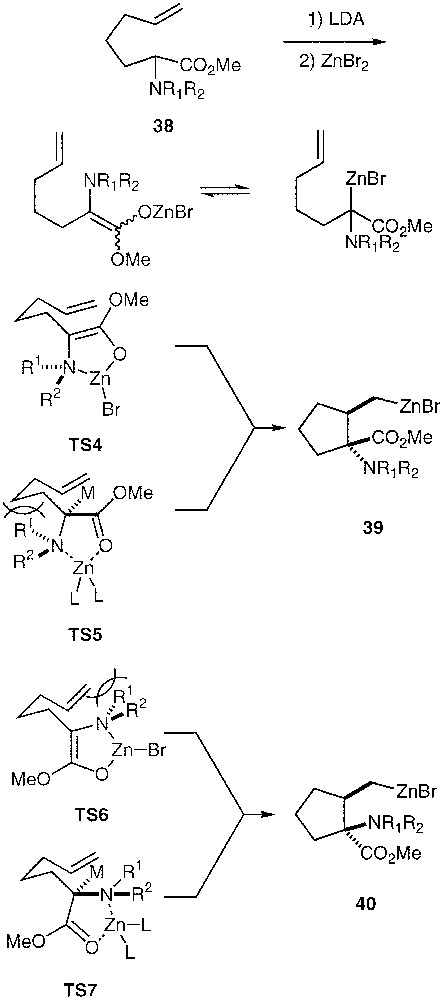
Carbocyclisation from aminoesters 38.
Compounds 38a, b and c were prepared through alkylation of the lithium enolate derived from the corresponding N,N-disubstituted amino methyl acetates 41a, b, c with 4-iodopentene in 75, 61 and 89% yield respectively (Fig. 11). Standard deprotonation of 38a, b, c with LDA followed by zinc bromide addition gave cyclised products 42a, b, c in 80, 36 and 19% unoptimized yields after hydrolysis and as a mixture of two diastereomers (based on 1H and 13C NMR analysis) in the 80:20 ratio. Reaction of the formed metalated cyclopentane derivative from 42a with allylbromide in the presence of copper cyanide gave the cyclopentanyl aminoester 43 in 58% yield and as a single isolated diastereomer.
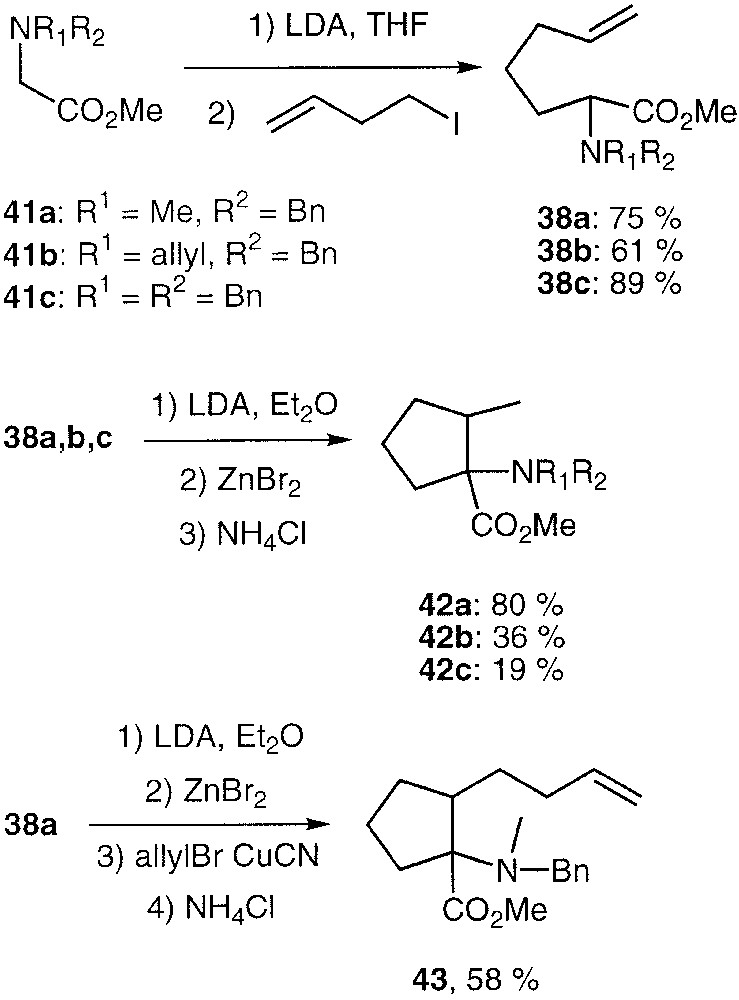
Stereoselective access to 1-amino-1-carbomethoxy-2-methyl cyclopentanes.
The relative stereochemistries of the compound 43 and of the major diastereomers of 42a, b, c could not been determined by any NMR experiment. However, reaction of the major diastereomer of 42a with an excess of p-tolyl Grignard reagent afforded the tertiary carbinol 44, which could be analysed through X-ray crystallographic studies. The relative stereochemistry was found to be cis (Fig. 12). We conclude that although being an α-aminoester, compound 38a has cyclised through a C-metalated enolate-ene reactive intermediate (see 40 from TS7 in Fig. 10), as in the case of β-(N-allyl)-aminoesters and, presumably, other α-(N-homoallyl)-aminoesters. It should be noted that this methodology affords a straightforward stereoselective route to 1-amino-1-carbomethoxy-2-methyl cyclopentanes difficult to prepare by other methods. We are currently working in our laboratory to enhance the yields and further explore the scope and limitations of this new promising methodology.

Stereochemical determination of cyclopentane 42a and 44.
4 Experimental Part
4.1 General remarks
Experiments involving organometallics were carried out in dried glassware under a positive pressure of dry nitrogen. Liquid nitrogen was used as a cryoscopic fluid. A round bottom flask equipped with an internal thermometer, a septum cap, a nitrogen inlet and a mechanic or magnetic stirring were used; THF was freshly distilled from sodium–benzophenone ketyl prior to use; zinc bromide (98%) was purchased from Aldrich. It was melted under dry nitrogen and, immediately after cooling down at room temperature, was dissolved in anhydrous THF; flash column chromatographic separations were carried out over Merck silica gel 60 (0.015–0.040 mm); 1H NMR and 13C NMR spectra were recorded on a Bruker AC 200 (respectively 200 and 50.3 MHz). Chemical shifts are reported in δ relative to an internal standard of residual chloroform (δ = 7.27 for 1H NMR and δ = 77.1 for 13C NMR); IR spectra were recorded with a Perkin-Elmer 1420 spectrophotometer; elemental analyses were performed by the ‘Service de microanalyses’ of the University Pierre-et-Marie-Curie (Paris-6).
4.2 General preparation of compounds 38
In a 250-ml round bottom four necked flask is cooled to – 78°C commercial LDA (36 mmol, 18 ml of a 2-M solution in n-heptane/THF) in THF (12 ml) and the methyl N-benzyl-N-alkylaminoacetate 41 (30 mmol) in THF (50 ml) is dropwise added. After stirring for 3 h at – 78 °C, 5-iodopent-1-ene (7.05 g, 36 mmol) in THF (50 ml) is added and the resulting yellow solution allowed to warm to room temperature. After stirring for 15 h, the reaction is hydrolysed with an aqueous solution of NH4Cl/NH3 (2:1) (100 ml). The resulting two layers are decanted, the aqueous one being extracted with ether (3 × 40 ml), the combined organic layers are washed with brine, dried over magnesium sulphate and the solvents evaporated under reduced pressure to give, after flash chromatography on silica gel, the title compounds 38.
4.2.1 Methyl 2-(N-benzyl-N-methyl)-amino-hept-6-enoate 38a
From aminoester 41a (5.86 g, 75%). IR (film, KBr): cm–1 3020, 2790, 1730, 1635; 1H NMR (CDCl3, 200 M Hz): δ 7.32–7.23 (m, 5H), 5.85–5.75 (m, 1H), 5.03–4.94 (m, 2H), 3.78 and 3.68 (AB system, 2H, J = 13.6 Hz), 3.73 (s, 3H), 3.31 (t, 1H), 2.26 (s, 3H), 2.09–2.03 (m, 2H), 1.78–1.71 (m, 2H), 1.57–1.53 (m, 1H), 1.45–1.41 (m, 1H); 13C NMR (CDCl3, 50 M Hz): δ 173.6, 140.0, 138.9, 129.1, 128.6, 127.3, 115.1, 65.8, 58.9, 51.3, 38.1, 33.8, 29.4, 25.9. Elemental analysis calcd (%) for C16H23NO2: C 73.53, H 8.87, N 5.36; found C 73.50, H 8.92, N 5.23.
4.2.2 Methyl 2-(N-allyl-N-benzyl)-amino-hept-6-enoate 38b
From aminoester 41b (5.25 g, 61%). 1H NMR (CDCl3, 200 M Hz): δ 7.31–7.18 (m, 5H), 5.89–5.64 (m, 2H), 5.24–4.90 (m, 4H), 3.94 and 3.49 (AB system, 2H, J = 14 Hz), 3.69 (s, 3H), 3.42–3.29 (m, 2H), 3.05 (ABX system, 1H, J = 7.86 Hz, J ́ = 14.26 Hz), 2.03–1.92 (m, 2H), 1.75–1.60 (m, 2H), 1.56–1.25 (m, 2H). 13C NMR (CDCl3, 50 M Hz): δ 173.96, 140.17, 138.70, 136.90, 128.88, 128.41, 127.07, 117.38, 114.92, 61.32, 54.55, 53.69, 51.17, 33.54, 29.76, 25.62. Elemental analysis calcd (%) for C18H25NO2: C 75.22, H 8.77, N 4.87; found C 75.20, H 8.85, N 4.82.
4.2.3 Methyl 2-N,N-dibenzylamino-hept-6-enoate 38c
From aminoester 41c (9.00 g, 89%). IR (film, KBr): cm–1 3060, 3030, 2950, 2840, 1730, 1640, 1600, 1490, 1450, 1430, 1360, 1200, 1140, 1075, 1630, 990, 915, 820, 780, 745, 700; 1H NMR (CDCl3, 200 M Hz): δ 7.40–7.20 (m, 10H), 5.80–5.70 (m, 1H), 4.98–4.92 (m, 2H), 3.95 et 3.53 (AB system, 4H, J = 14 Hz), 3.78 (s, 3H), 3.35 (dd, 1H, J = 10 Hz, J ́ = 6.6 Hz), 1.93 (q, 2H, J = 7 Hz), 1.84–1.67 (m, 2H), 1.63–1.52 (m, 1H), 1.40–1.30 (m, 1H); 13C NMR (CDCl3, 50 M Hz): δ 173.68, 139.84, 138.60, 129.00, 128.39, 127.13, 114.87, 60.62, 54.65, 51.15, 33.38, 29.09, 25.44. Elemental analysis calcd (%) for C22H27NO2: C 78.30, H 8.06, N 4.15; found C 78.14, H 8.13, N 4.08.
4.3 General procedure for the carbocyclisation to the compounds 42
In a 100-ml round bottom four-necked flask is cooled down to –40°C a solution of substituted aminoester 38 (2 mmol) in anhydrous THF (35 ml). Commercial LDA (10 mmol, 5 ml of a 2-M solution in n-heptane/THF) is dropwise added and the mixture is allowed to warm to 10 °C for 10 min. After cooling down to –40 °C, ZnBr 2 (8 mmol, 8 ml of a 1-M solution in THF) is added and the reaction mixture is allowed to warm to room temperature and stirred 20 min for 38a and 3 h for 38b. After hydrolysis with an aqueous solution of NH4Cl/NH3 (2:1) (50 ml), the aqueous layer is extracted with ether (3 × 20 ml). The combined organic layers are washed with brine, dried over magnesium sulphate and the solvents evaporated under reduced pressure to give, after chromatography on silica gel, the title compounds 42. Only the major diastereomer for 42a and 42b is described below. The compound 42c could not be separated from the starting material.
4.3.1 Methyl 1-(N-benzyl-N-methylamino)-2-methyl-cyclopentanecarboxylate 42a
From compound 38a (80%, 0.42 g). IR (film, KBr): cm–1 2960, 2800, 1720, 1495, 1450, 1375, 1235, 1190, 1065, 910, 735, 700; 1H NMR (CDCl3, 200 M Hz): δ 7.36–7.30 (m, 5H), 3.76 (s, 3H), 3.60 (s, 2H), 2.69–2.64 (m, 1H), 2.42–2.37 (m, 1H), 2.09 (s, 3H), 1.82–1.76 (m, 3H), 1.62–1.58 (m, 1H), 1.51–1.45 (m, 1H), 1.03 (d, 3H, J = 7.08 Hz); 13C NMR (CDCl3, 50 MHz): δ 174.0, 140.6, 128.30, 128.1, 127.7, 76.8, 58.0, 51.1, 37.8, 37.7, 31.7, 31.05, 20.2, 15.8; Elemental analysis calcd (%) for C16H23NO2: C 73.53, H 8.87, N 5.36; found C 74.23, H 9.02, N 5.02.
4.3.2 Methyl 1-(N-allyl-N-benzylamino)-2-methyl-cyclopentanecarboxylate 42b
From compound 38b (36%, 0.21 g). IR (film, KBr): cm–1 2960, 1720, 1640, 1600, 1490, 1450, 1375, 1330, 1280, 1235, 1190, 1100, 1075, 1040, 1030, 1000, 975, 920, 795, 735, 700; 1H NMR (CDCl3, 200 M Hz): δ 7.37–7.13 (m, 5H), 5.81–5.60 (m, 1H), 4.99–4.83 (m, 2H), 3.90 and 3.73 (AB system, 2H, J = 16 Hz), 3.71 (s, 3H), 3.30 (ABX system, 1H, J = 4.92 Hz, J ́ = 15.51 Hz), 3.05 (ABX system, 1H, J = 7.86 Hz, J ́ = 14.76 Hz), 2.74–2.60 (m, 1H), 2.26–2.18 (m, 1H), 1.78–1.25 (m, 5H), 1.01 (d, 3H, J = 7.12 Hz); 13C NMR (CDCl3, 50 M Hz): δ 175.35, 141.89, 136.59, 128.04, 128.01, 126.41, 116.40, 77.2, 55.20, 54.92, 51.24, 38.41, 31.96, 30.76, 20.34, 16.35; Elemental analysis calcd (%) for C18H25NO2: C 75.22, H 8.77, N 4.87; found C 75.51, H 8.15, N 4.57.
4.4 Methyl 1-(N-benzyl-N-methylamino)-2-[but-3-enyl]-cyclopentanecarboxylate 43
In a 100-ml round bottom three-necked flask is cooled down to – 78 °C a solution of LDA (5 mmol, 2.5 ml of a 2-M solution in n-heptane/THF) in THF (5 ml). The substituted aminoester 38a in THF (5 ml) is dropwise added, the mixture stirred for 30 min at –78 °C and allowed to warm to room temperature for 2 h. ZnBr2 (5 mmol, 5 ml of a 1-M solution in THF) is added and the resulting mixture stirred for 4 h. The mixture is cooled down to –40 °C and CuCN (0.445 g, 5 mmol) is added in one portion. The mixture is warmed to room temperature, cooled down to –40 °C and allyl bromide 0.5 ml, 5 mmol: in THF (2 ml) is added. The reaction mixture is allowed to warm to room temperature and stirred overnight. The reaction is hydrolysed with an aqueous solution of NH4Cl/NH3 (2:1) (30 ml), the aqueous layer is extracted with ether (3 × 10 ml), the combined organic layers washed with brine, dried over magnesium sulphate and the solvents evaporated under reduced pressure to give, after flash chromatography on silica gel, the title compound (0.291 g, 58%). IR (film, KBr): cm–1 3060, 3020, 2960, 2800, 1720, 1635, 1600, 1495, 1450, 1360, 1280, 1195, 1170, 1120, 1070, 1030, 995, 910, 790, 735, 700; 1H NMR (CDCl3, 200 M Hz): δ 7.35–7.19 (m, 5H), 5.92–5.72 (m, 1H), 5.08–4.91 (m, 2H), 3.75 (s, 3H), 3.62 and 3.49 (AB system, 2H, J = 14 Hz), 2.47–2.37 (m, 2H), 2.25–2.15 (m, 1H), 2.05 (s, 3H), 1.99–1.04 (m, 8H); 13C NMR (CDCl3, 50 M Hz): δ 173.07, 140.49, 139.07, 129.32, 129.23, 126.78, 114.75, 76.56, 57.94, 51.15, 42.73, 37.64, 32.9, 32.22, 27.43, 27.15, 20.17; Elemental analysis calcd (%) for C19H27NO2: C 75.71, H 9.03, N 4.65; found C 76.02, H 9.32, N 4.48.
4.5 [1-(N-benzyl-N-methylamino)-2-methyl-cyclopentyl]di-p-tolyl-methanol 44
A solution of 4-methyl phenyl bromide (1.44 g, 8.44 mmol) in anhydrous ether (20 ml) is cooled down to –80 °C and tBuLi (16.38 mmol, 9.6 ml of a 1.7-M solution in pentane) is dropwise added. The resulting mixture is stirred at 0 °C for 1 h. The compound 42a (0.55 g, 2.11 mmol) in anhydrous ether (3 ml) is dropwise added at –5 °C and the reaction mixture allowed to warm to room temperature and stirred overnight. After hydrolysis with an aqueous solution of NH4Cl/NH3 (2:1) (20 ml), the aqueous layer is extracted with ether (3 × 10 ml), the combined organic layers washed with brine, dried over magnesium sulphate and the solvents evaporated under reduced pressure to give, after flash chromatography, the title compound 44 (0.556 g, 64%) as a white solid. mp: 110–113 °C; IR (film, KBr): cm–1 3050, 2950, 1500, 1450, 1265, 1040, 1015, 945, 895, 810, 740, 705; 1H NMR (CDCl3, 200 M Hz): δ 7.79–7.66 (m, 4H, H; 7.29–7.13 (m, 5H, H); 7.05–6.97 (m, 4H), 6.45 (s, 1H), 3.87 and 3.59 (AB system, 2H, J = 13.54 Hz), 2.62–2.43 (m, 2H), 2.39–1.26 (m, 5H), 2.27 (s, 3H), 2.23 (s, 3H), 2.04 (s, 3H), 1.26 (d, 3H); 13C NMR (CDCl3, 50 M Hz): δ 147.37, 144.32, 140.97, 135.62, 128.79, 128.66, 128.42, 127.08, 126.79, 126.63, 81.13, 79.95, 58.52, 42.02, 38.97, 33.12, 32.80, 29.99, 23.12, 21.09, 15.84; Elemental analysis calcd (%) for C29H35NO: C 84.22, H 8.53, N 3.39; found C 83.80, H 8.65, N 3.12.
Supplementary material
The supplementary material has been sent to the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK, as supplementary material No. 196065 (6 pages) and can be obtained by contacting the CCDV/FIZ.
Acknowledgements
Thanks are due to Carine Guyard-Duhayon, from the ‘Laboratoire de chimie inorganique et matériaux moléculaires’, University Pierre-et-Marie-Curie (Paris-6) for X-ray crystallographic studies.