

1 Introduction
α-Coupled polythiophenes have unusual properties and have attracted substantial interest [1–3]. In particular, oligothiophenes are of major importance in the second order non-linear optic field because molecules containing the sequence electron donor-spacer-electron acceptor present a higher hyperpolarizability when a thienyl spacer replaces a classical phenyl group [4–7]. Thus, we developed an access to functionalized oligothiophenes using transition-metal-catalyzed coupling reactions [8]. This approach enabled an easy synthesis of well-defined oligothiophenes that could be readily functionalized. In particular, we intended to prepare ortho- and meta-disubstituted arenetricarbonylchromium complexes in order to study their influence on the optoelectronic properties of the molecules.
2 Results and discussion
In the first part of this paper, we will describe the synthesis of oligothiophenes using Stille coupling reaction. In the second part, we will show how to modify them with arenetricarbonylchromium complexes.
The preparation of the key synthons 2a and 2b is depicted in Fig. 1. Commercially available 2-(3-thienyl)ethanol 1 was quantitatively O-benzylated according to a standard procedure [9] and regiospecifically brominated at the 2 position using N-bromosuccinimide in DMF owing to an aromatic electrophilic substitution [10–11]. Stannylation firstly required lithium–halogen exchange at low temperature, followed by addition of trimethyl or tributylstannyl chloride. Two different stannyl chlorides were used in order to compare their reactivity in the subsequent coupling reactions (Fig. 1).
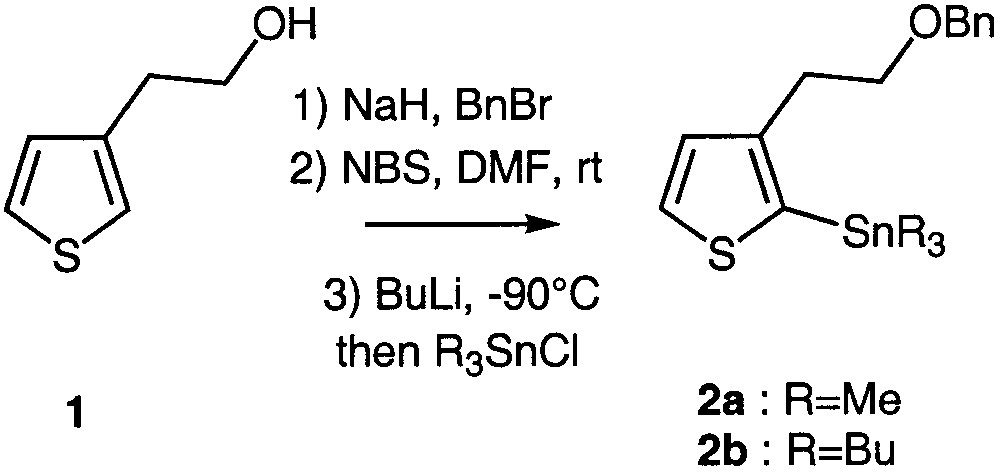
Preparation of the key building blocks 2a and 2b.
As trialkylstannylthiophenes exhibit low stability upon silica-gel-chromatography purification [12] and as they were pure enough for being used as raw material, they were directly involved in the coupling reactions without any purification (Fig. 2).
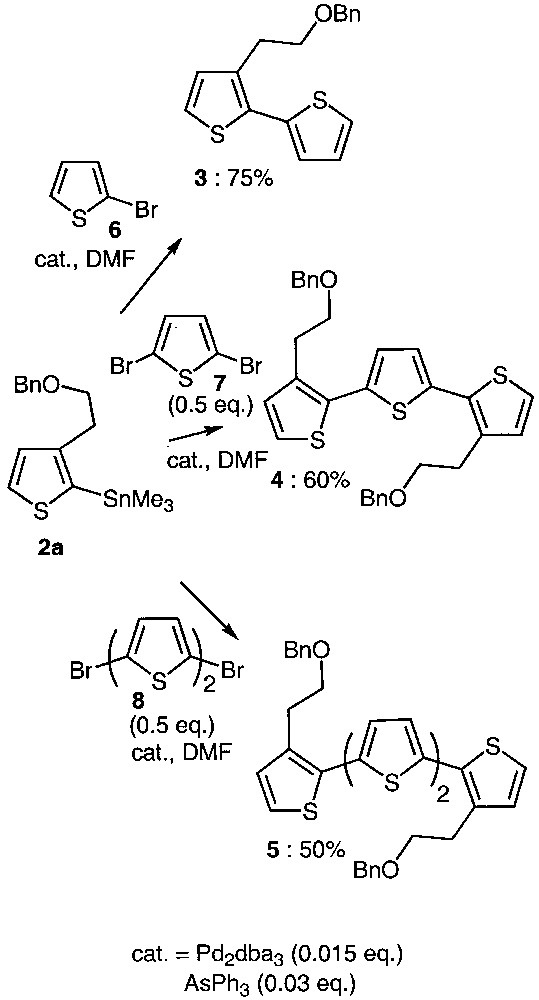
Synthetic approach to oligothiophenes 3–5.
Combination of Pd2dba3 as a catalyst precursor and AsPh3 as ligand was used to generate the catalytic species required for the Stille reaction [13–17]. As shown in Fig. 2, compound 2a reacted with the 2-bromothiophene 6, 2,5-dibromothiophene 7 as well as 5,5′-dibromobithiophene 8 in DMF under the above-mentioned catalytic conditions, affording oligothiophenes 3, 4 and 5 in reasonable yields. Optimizing the coupling conditions led to the use of low catalyst loading. Indeed, it has been determined that only 1.5% of palladium source and 3% of ligand are sufficient to ensure completion of the coupling reaction. Comparing the reactivity of tributyl and trimethylstannylthiophene proved that the reaction proceeds in comparable yields but is faster with 2a. According to this result, the trimethylstannyl derivative 2a was preferentially chosen for further reactions.
Moreover, we have shown that for the same reaction time (24 h), the temperature required for the coupling varies from room temperature to 153 °C. It is interesting to note that the 2,5-dibromothiophene 7 only reacts at high temperature. In contrast, 5,5′-dibromobithiophene 8 behaves like two independent bromothiophene units, and the reaction proceeds at room temperature in 50% yield.
Using this approach, compound 5 could be selectively and quantitatively brominated at the α position [18] and accordingly coupled with stannane 2a, affording sexithiophene 9 in 60% yield (Fig. 3). As already related for 5,5′-dibromobithiophene 8, the independent character of the 5 and 5′′′ bromide atoms allowed the reaction to be carried out at room temperature (Table 1).
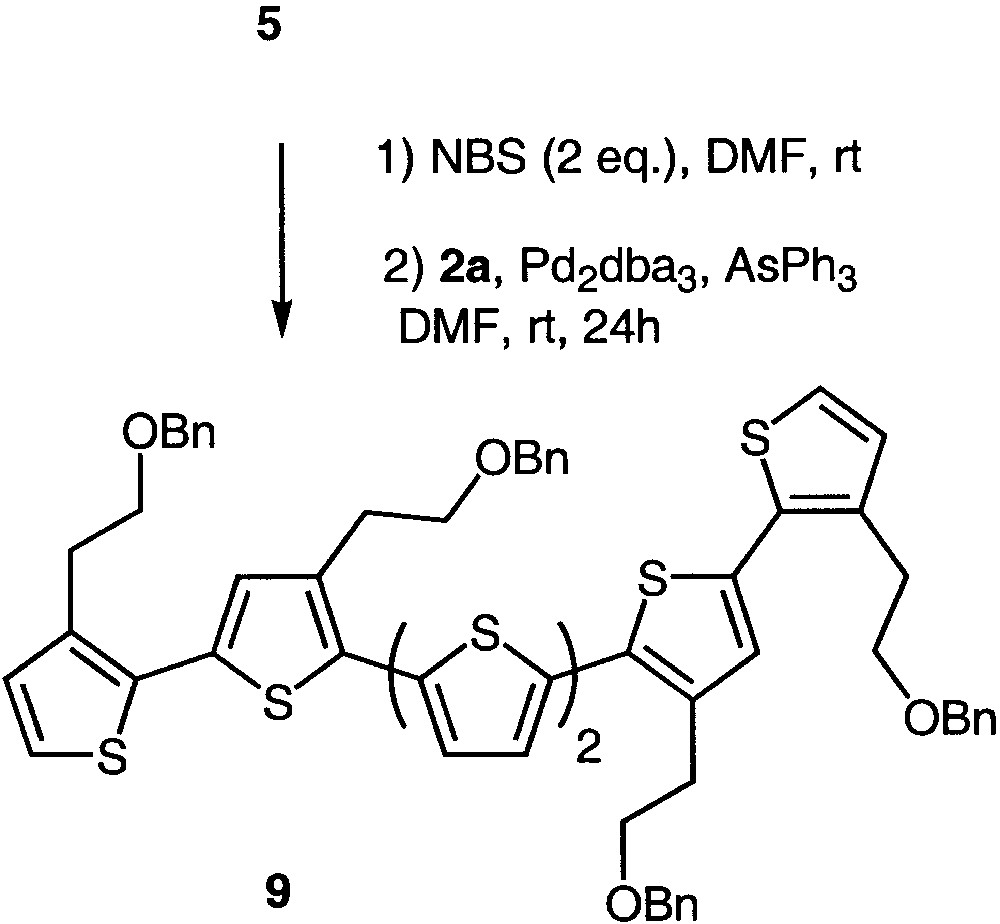
Synthesis of sexithiophene 9.
Summarized data for the coupling reactions
| Oligothiophene | Temperature | Yield (%) |
| 3 | 60 °C | 75 |
| 4 | 153 °C | 60 |
| 5 | rt | 50 |
| 9 | rt | 60 |
Having in hands an efficient strategy for the preparation of well-defined oligothiophenes, we then investigated an access to more elaborated systems. The synthetic approach that we chose involved the deprotection of the benzyl ether. Preliminary attempts were carried out using the more readily available bithiophene 3 as model compound.
While hydrogenolysis did not afford the expected alcohol 10, compound 3 was quantitatively de-benzylated during 10 min using iodotrimethylsilane at room temperature in freshly distilled dichloromethane (Fig. 4) [19].
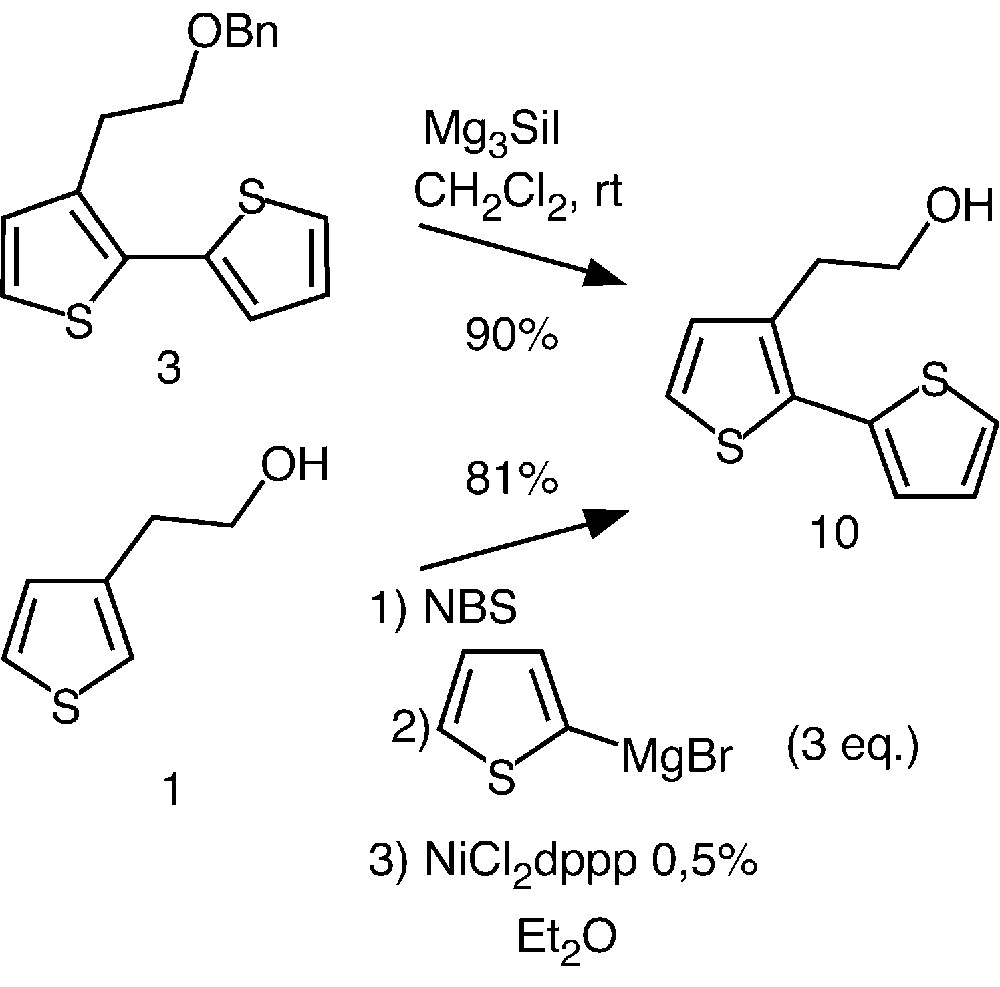
Preparation of compound 10 by two different pathways.
Alternatively, we also investigated a straightforward preparation of 10 based on the so-called Kumada coupling reaction [20–22]. This one-pot strategy involves bromination of thiophene 1 and coupling of the latter with 2-thienylmagnesium bromide in the presence of NiCl2dppp (dppp = 1,3-bis(diphenylphosphino)propane). Interestingly, this approach led to 10 in 81% yield. In contrast to the Stille reaction (Fig. 2), the new pathway rapidly completed in a few minutes and required an easy purification step. Thus, using this very efficient method, we were able to prepare compound 10 in gram-scale quantity, allowing further functionalization.
Considering our expertise in arene-chromium chemistry [23–24], we intended to link ortho- and meta-disubstituted arene-chromium complexes with planar chirality [25] to bithiophene 10 via an ipso substitution [26–33] (Fig. 5), in order to study the potential response of the resulting molecules in NLO [6].
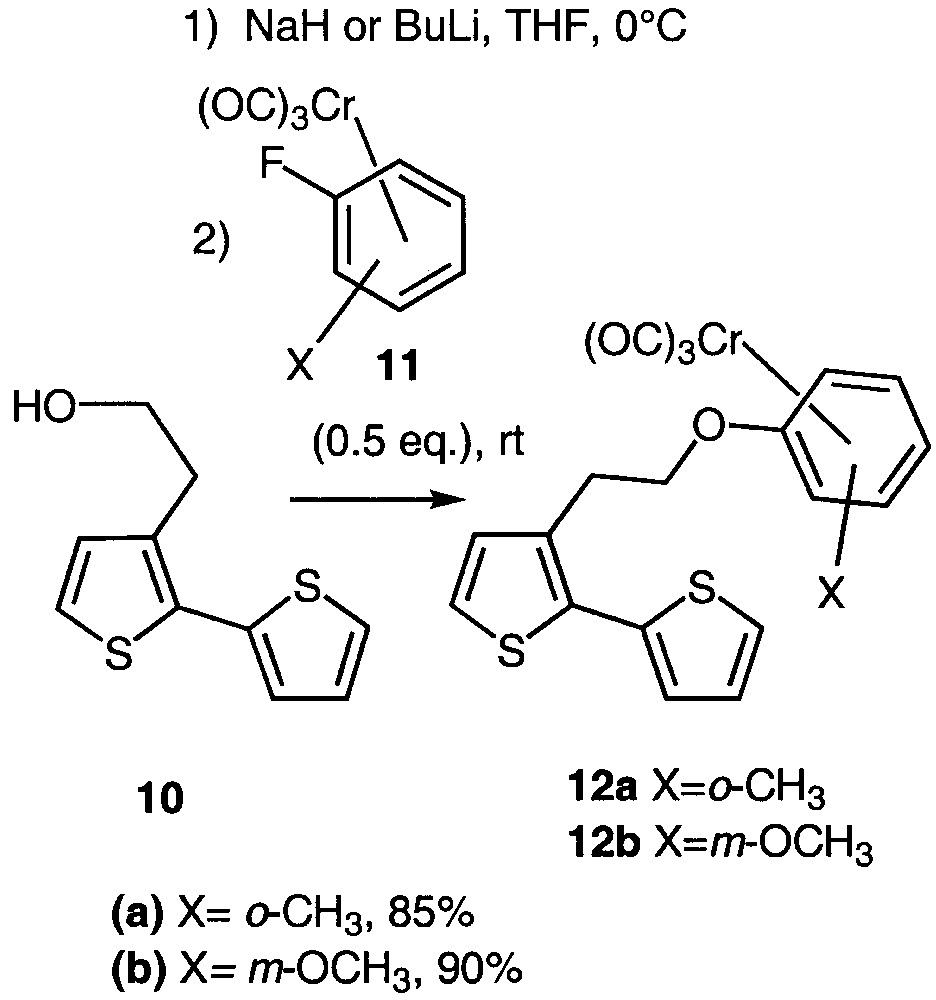
Ipso substitution leading to organometallic bithiophenes 12a and 12b.
The method involves an ipso nucleophilic aromatic substitution of the fluorine atom of ortho-fluorotoluene 11a or meta-fluoroanisole 11b chromium complexes [34] by the alcoholate derived from 10. Alcohol 10 could either be deprotonated with n-butyllithium or sodium hydride without noticeable difference in the chemical yield. In a first attempt, the use of equimolar amounts of alcoholate and arene-chromium afforded an unsatisfying 30% yield. Fortunately, the use of an excess of alcoholate (2 equiv) led to a huge improvement as the coupled compounds 12a and 12b were isolated in 85 and 90% yield respectively.
Noteworthy, 1H NMR data of compound 12a showed interesting features. The proton signals of the ethoxy chain in compound 12a exhibit the expected apparent triplet for Ha and Hb (Fig. 6a). However, the multiplicity of the signal corresponding to Hc and Hd appears as a doublet of doublet of doublet, due to the fact that they are diastereotopic because of the presence of the stereogenic ortho-disubstituted arenetricarbonylchromium moiety. Selective irradiation of the upfield triplet confirmed the presence of two diastereotopic protons (Fig. 6b).
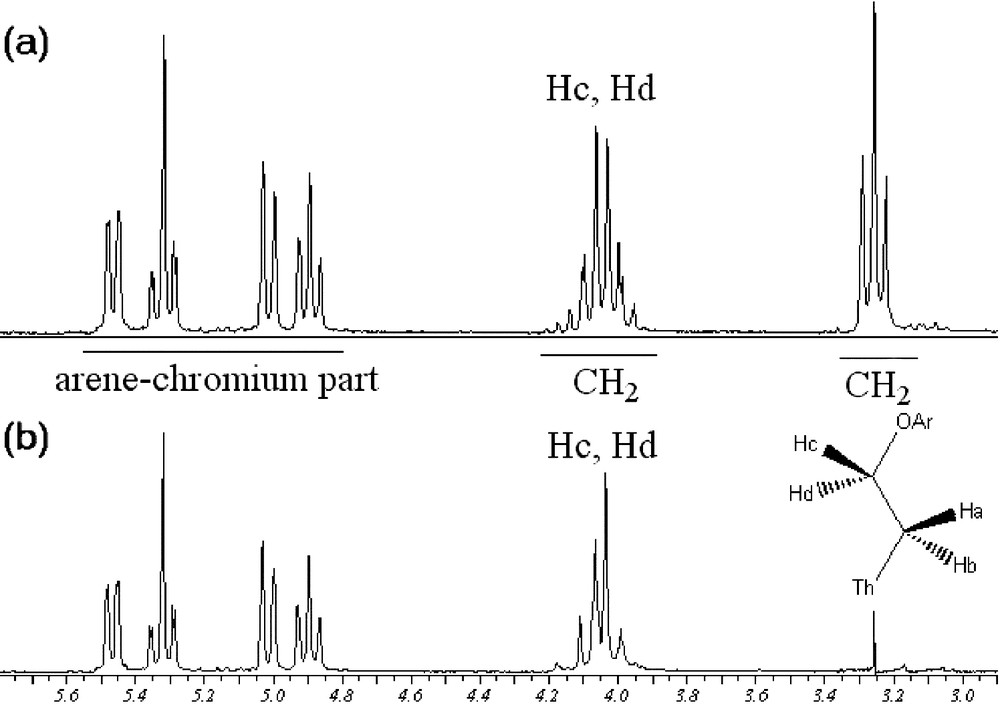
Irradiation of Ha and Hb, protons of 12a (Th = thiophene; Ar = arenetricarbonylchromium complexes).
Interestingly, the 1H NMR spectrum of complex 12b revealed two triplets, corresponding to the ethoxy chain. In comparison with 12a, the influence of the planar chirality on the multiplicity of the C2H4O unit can easily be explained by the fact that the diastereotopic Hc and Hd protons resonance frequencies are accidentally equal (isochronous).
It has been reported that in solution a substituted arenetricarbonylchromium complex can be considered to be in equilibrium between two conformations: the syn-eclipsed SE conformation with an (x) population and the anti-eclipsed AE conformation with an (1 – x) population (Fig. 7). Thus, if D is an electron-donor substituent, the major conformer is the SE one and if D is an electron-withdrawing group, the major conformer is the AE one [36].
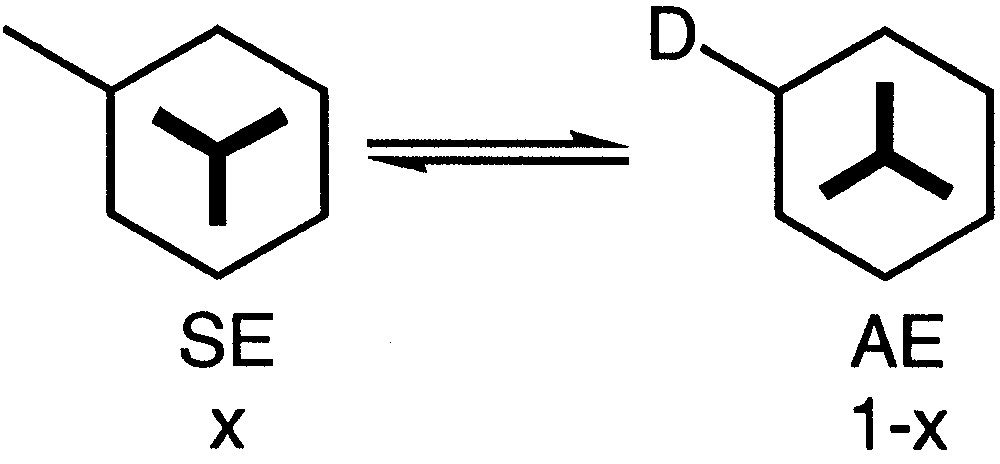
Equilibrium between two conformers SE and AE.
The conformation of the Cr(CO)3 tripod of complexes 12a and 12b can be studied in solution by analysing the 1H NMR spectra. Indeed, in both cases the conformation of the major conformer was eclipsed with respect to the alkoxy group according to the data gathered in Table 2. The resonances of these eclipsed protons H3 and H5 occur, as expected, at high frequency [35]. The largest difference of chemical shift was 0.57 in the case of complex 12a and reached 0.77 in the case of complex 12b. Thus, it was possible to calculate the population x of the major isomer (Table 2) [36–40].
Selected 1H NMR data of complexes 12a and 12b. a δH3 – δH4 = 0.57; b δH5 – δH4 = 0.43; c δH5 – δH6 = 0.31; d δH5 – δH4 = 0.77; e δHi – δHi–1 = (2 x – 1) Δδmax, see refs [36–40]. The maximum difference Δδmax used in this reference is 0.84 ppm in CDCl3, which represents the difference of chemical shifts between the eclipsed 1,3,5-tri-methylbenzene and the anti eclipsed 1,3,5-tri-tertio-butylbenzene arenetricarbonylchromium complexes.
| Complex | H2 | H3 | H4 | H5 | H6 | xe |
| — | 5.46 | 4.89a | 5.32b | 5.01c | 68% | |
| 5.13 | — | 4.78d | 5.55 | 4.78 | 92% |
In conclusion, we have described a selective and efficient small-scale method for accessing bi-, tri-, quater- and sexithiophenes. The nickel-catalyzed method allowed us to prepare bithiophene 10 in quantitative yield from commercially available compounds. We also reported the first examples of arene-chromium complexes associated to oligothiophenes prepared in high yield by ipso-nucleophilic aromatic substitution SNAr of the fluorine atom of o-fluorotoluene or m-fluoroanisole chromium complexes. Further insights on the non-linear properties of the molecules are currently in progress.
3 Experimental part
3.1 General information
2-(3-thienyl)ethanol, 2-bromothiophene, Pd2dba3, N-bromosuccinimide were purchased from ACROS. Tributylstannyl chloride, AsPh3, benzyl bromide, are available from Aldrich Chemical Co. Organic solvents were dried using standard procedures. Additionally, DMF was degassed with N2 bubbling. Analysis apparatus: NMR spectrum on Brüker AC200 and ARX400, Infrared spectrum on Nicolet-Avatar 320 FT-IR, UV spectrum on UVIKON 923, MS/HRMS on Applied Biosystem Voyager DE-STR MALDI-TOF MS.
3.2 Bromination of 3-(2-benzyloxy-ethyl)-thiophene
To a solution of 7.012g (32mmol) of benzyloxy-2-(3-thienyl)ethanol in 100 ml of DMF was added dropwise during 1 h a solution of 5.77 g (32 mmol) of N-bromosuccinimide in 50 ml of DMF sheltered from light, the mixture was stirred overnight. The mixture was poured into 200 ml of water and extracted three times with diethylether. The organic layer was treated with a 10% solution of KOH and washed with water. The organic layer was dried over MgSO4 and evaporated under reduced pressure. The crude product (8.905 g) was distilled under vacuum (E1 = 110 °C, yellow oil, 7.132 g, 75%). The crude product can be also purified by silica-gel-column chromatography using a mixture of cyclohexane/dichloromethane: 80:20 (yield = 75%). 1H NMR (200 MHz, CDCl3) δ, ppm: 7.33 (m, 5H, Har); 7.20 (d, 1H, H5, J = 5.9 Hz); 6.88 (d, 1H, H4, J = 5.9 Hz); 4.54 (s, 2H, Hbenz); 3.67 (t, 2H, Halk, J = 6.9 Hz); 2.92 (t, 2H, Halk, J = 6.9 Hz). 13C NMR (50 MHz, CDCl3) δ, ppm: 138.4; 128.9; 128.5; 127.7; 125.5; 110.1; 73.0; 69.3; 30.2. Anal. calc. for C13H13BrOS: C 52.53, H 4.41; found: C 52.42, H 4.43.
3.3 Stannylation of 3-(2-benzyloxy-ethyl)-2-bromo-thiophene, synthesis of 2a
To a solution of 300 mg (1 mmol) of 3-(2-benzyloxy-ethyl)-2-bromo-thiophene in anhydrous diethylether at –90 °C was added dropwise 630 μl (1 mmol) of a solution of n-BuLi (1.6 M) in hexane. After 10 min, 1 ml (1 mmol) of a solution of Me3SnCl (1M) in THF was added dropwise and the mixture was stirred for 1h at –90°C. Then, water was added directly at –90°C and the mixture was allowed to warm up at room temperature, then extracted with diethylether, washed with water, dried over MgSO4 and evaporated under reduced pressure. The [3-(2-benzyloxy-ethyl)-thiophen-2-yl]-trimethyl-stannane 2a was obtained in 90% yield and 10% yield of debrominated thiophene measured by NMR were recovered. The crude product is used without further purification. 1H NMR (200 MHz, CDCl3) δ, ppm: 7.5 (d, 1H, H5, J = 4.9 Hz); 7.32 (m, 5H, Har); 7.13 (d, 1H, H4, J = 4.9 Hz); 4.54 (s, 2H, Hbenz); 3.67 (t, 2H, Halk, J = 6.9 Hz); 2.92 (t, 2H, Halk, J = 6.9 Hz). 13C NMR (50 MHz, CDCl3) δ, ppm: 146.6; 138.6; 133.3; 131.0; 130.0; 128.6; 127.9; 127.8; 73.2; 71.5; 33.2; –7.6.
3.4 General procedure: coupling reactions
To a solution of [3-(2-benzyloxy-ethyl)-thiophen-2-yl]-trimethyl-stannane 2a in DMF, was added a solution of brominated thiophene (1 or 0.5 equiv according to the coupling reaction), Pd2dba3 (0.015 equiv), AsPh3 (0.03 equiv) in DMF. The mixture was stirred at the appropriated temperature (Table 1). After 24 h, the mixture was poured into water and extracted three times with diethylether. The combined organic phases were dried over MgSO4 and evaporated under reduced pressure.
3.5 Synthesis of 3-(2-benzyloxy-ethyl)-[2,2′]bithiophenyl (3)
Compound 3 was prepared according to the general procedure at 60 °C. The crude product can be also purified by silica-gel-column chromatography using a mixture of cyclohexane/dichloromethane: 70:30 then 60:40 (yellow oil, 75% yield). 1H NMR (400 MHz, CDCl3) δ, ppm: 7.32 (m, 5H, Har); 7.30 (dd, 1H, H5′, J = 5.2 Hz, J = 1.2 Hz); 7.19 (d, 1H, H5, J = 5.2 Hz); 7.12 (dd, 1H, H3′, J = 3.6 Hz, J = 1.2 Hz); 7.05 (dd, 1H, H4′, J = 5.2 Hz, J = 3.6 Hz); 7.00 (d, 1H, H4, J = 5.2 Hz); 4.53 (s, 2H, Hbenz); 3.71 (t, 2H, Halk, J = 7.1 Hz); 3.10 (t, 2H, Halk, J = 7.1 Hz). 13C NMR (100 MHz, CDCl3) δ, ppm: 138.4; 135.8; 135.6; 131.8; 130.1; 128.4; 127.6; 127.5; 127.4; 126.3; 125.6; 124.0; 72.9; 70.1; 29.6. UV/Visible (CHCl3): λ (nm) = 293, ϵ = 8614 cm–1 mol–1 l. HRMS (MALDI-TOF) m/z: calculated for C17H16OS2 (+Na) = 323.0549; found (+Na) = 323.0535.
3.6 Synthesis of 3,3′′-bis-(2-benzyloxy-ethyl)-[2,2′;5′,2′′]terthiophene (4)
Compound 4 was prepared according to the general procedure at 153 °C. The crude product was chromatographed on silica gel using a mixture of cyclohexane/dichloromethane: 65:35, 55:45 and then 45:55 (orange oil, 60% yield). 1H NMR (200 MHz, CDCl3) δ, ppm: 7.30 (m, 10H, Har); 7.20 (d, 2H, H5, H5″, J = 5.1 Hz); 7.05 (s, 2H, H3′, H4′); 7.00 (d, 2H, H4, H4′′, J = 5.1 Hz); 4.53 (s, 4H, Hbenz); 3.72 (t, 4H, Halk, J = 6.9 Hz); 3.11 (t, 4H, Halk, J = 6.9 Hz). 13C NMR (50 MHz, CDCl3) δ, ppm: 138.4; 136.0; 135.9; 131.6; 130.3; 128.4; 127.7; 126.7; 124.2; 73.0; 70.2; 29.8. UV/visible (CHCl3): λ (nm) = 334, ϵ = 17 000 cm–1 mol–1 l. HRMS (MALDI-TOF) m/z: calculated for C30H28O2S3 = 516.1423; found = 516.1246.
3.7 Synthesis of 3,3′′′-bis-(2-benzyloxy-ethyl)-[2,2′;5′,2′′;5′′,2′′′]quaterthiophene (5)
Compound 5 was prepared according to the general procedure at room temperature. The crude product can be also purified by silica-gel-column chromatography using a mixture of cyclohexane/dichloromethane: 65:35 then 55:45 and then 45:55 (orange solid, 50% yield). 1H NMR (200 MHz, CDCl3) δ, ppm: 7.34 (m, 10H, Har); 7.20 (d, 2H, H5, H5′′′, J = 5.2 Hz); 7.01 (d, 2H, H4, H4′′′, J = 5.2 Hz); 7.12 (d, 2H, H4′, H3″, J = 3.9 Hz); 7.05 (d, 2H, H3′, H4″, J = 3.9 Hz); 4.56 (s, 4H, Hbenz); 3.75 (t, 4H, Halk, J = 6.9 Hz); 3.14 (t, 4H, Halk, J = 6.9 Hz). 13C NMR (50 MHz, CDCl3) δ, ppm: 138.5; 137.1; 135.9; 135.1; 131.7; 130.5; 128.6; 127.8; 127.1; 124.3; 124.2; 73.1; 70.2; 30.0. UV/visible (CHCl3): λ (nm) = 377, ϵ = 28 000 cm–1 mol–1 l. HRMS (MALDI-TOF) m/z: calculated for C34H30O2S4 = 598.1123; found = 598.1044.
3.8 Bromination of 3,3′′′-bis-(2-benzyloxy-ethyl)-[2,2′;5′,2′′;5′′,2′′′]quaterthiophene; preparation of 3′′′-(2-benzyloxy-ethyl)-5,5′′′-dibromo-3-(2-phenoxy-ethyl)-[2,2′;5′,2′′;5′′,2′′′]quaterthiophene
According to the bromination of 2, the workup was done on 50 mg (0.083 mmol) of 5 and 30 mg of NBS (2 equiv) (orange solid, 60mg, 95% yield). 1H NMR (200 MHz, CDCl3) δ, ppm: 7.31 (m, 10H, Har); 7.07 (d, 2H, H4′, H3′′, J = 3.9 Hz); 6.98 (d, 2H, H3′, H4′′, J = 3.9 Hz); 6.95 (s, 2H, H4, H4′′′); 4.53 (s, 4H, Hbenz); 3.68 (t, 4H, Halk, J = 6.4 Hz); 3.03 (t, 4H, Halk, J = 6.4 Hz). 13C NMR (50 MHz, CDCl3) δ, ppm: 138.2; 137.2; 136.6; 133.7; 133.1; 128.5; 127.7; 127.4; 124.2; 110.9; 73.1; 69.7; 29.8. HRMS (MALDI-TOF) m/z: calculated for C34H28Br2O2S4 = 755.9314; found = 755.9266.
3.9 Synthesis of 3,4′,3′′′′,3′′′′′-tetrakis-(2-benzyloxy-ethyl)-[2,2′;5′,2′′;5′′,2′′′;5′′′,2′′′′;5′′′′, 2′′′′′]sexithiophene (9)
Compound 9 was prepared according to the general procedure at room temperature. The crude product can be also purified by silica-gel-column chromatography using a mixture of cyclohexane/dichloromethane: 50:50 (orange oil, 60% yield). 1H NMR (400 MHz, CDCl3) δ, ppm: 7.35 (m, 20H, Har); 7.20 (d, 2H, H5, H′′′′, H4′′′′); 7.02 (d, 2H, H4, H4′′′′, J = 5.1 Hz); 4.58 (s, 4H, Hbenz); 4.56 (s, 4H, Hbenz); 3.78 (t, 4H, Halk, J = 7.1 Hz); 3.76 (t, 4H, Halk, J = 7.1 Hz); 3.16 (t, 8H, Halk, J = 7.1 Hz). 13C NMR (100 MHz, CDCl3) δ, ppm: 138.5; 138.4; 137.1; 136.3; 135.9; 134.7; 134.3; 131.7; 131.7; 130.4; 129.5; 128.5; 127.7; 126.9; 126.8; 124.2; 124.1; 73.1; 70.2; 70.0; 30.1; 29.9; 29.8. UV/visible (CHCl3): λ (nm) = 411; ϵ = 27 292 cm–1 mol–1 l. HRMS (MALDI–TOF) m/z: calculated for C60H54O4S6 = 1030.2316; found = 1030.2341.
3.10 Synthesis of 2-[2,2′]bithiophenyl-3-yl-ethanol (10) from 3-(2-benzyloxy-ethyl)-[2,2′]bithiophenyl (3)
To a 2-M solution of 3 (50 mg, 0.166 mmol) in freshly distilled dichloromethane (84 μl) was added dropwise 32 μl (0.166mmol) of iodotrimethylsilane. The reaction was stirred under nitrogen for 1 hour. The mixture is poured into water, extracted with dichloromethane, dried over Na2SO4 and evaporated under reduced pressure. The crude product can be also purified by silica-gel-column chromatography using dichloromethane (colourless oil, 32mg, 90% yield). 1H NMR (200 MHz, CDCl3) δ, ppm: 7.28 (dd, 1H, H5′, J = 5.2 Hz, J = 1.5 Hz); 7.19 (d, 1H, H5, J = 5.2 Hz); 7.15 (dd, 1H, H3′, J = 3.7 Hz, J = 1.5 Hz); 7.04 (dd, 1H, H4′, J = 5.2 Hz, J = 3.7 Hz); 6.96 (d, 1H, H4, J = 5.2 Hz); 3.81 (t, 2H, Halk, J = 6.9 Hz); 3.00 (t, 2H, Halk, J = 6.9 Hz). 13C NMR (50 MHz, CDCl3) δ, ppm: 135.7; 135.4; 132.3; 130.1; 127.6; 126.6; 125.8; 124.4; 62.8; 32.4. HRMS (MALDI–TOF) m/z: calculated for C10H10OS2 (+Na) = 233.0065; found (+Na) = 233.0059. Anal. calc. for C10H10OS2: C 57.11, H 4.79; found: C 56.95, H 4.99.
3.11 Synthesis of 2-[2,2′]bithiophenyl-3-yl-ethanol (10) from 2-(3-thienyl)-ethanol (1)
Brominated thienylethanol was prepared according to the bromination of 2 from 2-(3-thienyl)-ethanol (2 g, 15.6 mmol) in 50 ml of DMF and NBS (2.387 g, 15.6 mmol) to yield 2.536 g (93%) of a colourless oil. A solution of 2.99 g (18 mmol) of 2-bromothiophene 6 and 446 mg of magnesium in anhydrous diethylether was stirred until complete dissolution of the metal; the brominated thienylethanol (1.27 g, 6.13 mmol) was added dropwise to the Grignard reagent. 0.5% of the nickel catalyst (17mg) was then added to the reaction. The reaction was stirred under N2 for 30 min. The mixture was poured into water, extracted with diethylether, dried over MgSO4 and evaporated under reduced pressure. The crude product can be also purified by silica-gel-column chromatography using dichloromethane (colourless oil, 1.16g, 90% yield). 1H NMR (200 MHz, CDCl3) δ, ppm: 7.28 (dd, 1H, H5′, J = 5.2 Hz, J = 1.5 Hz); 7.19 (d, 1H, H5, J = 5.2 Hz); 7.15 (dd, 1H, H3′, J = 3.7 Hz, J = 1.5 Hz); 7.04 (dd, 1H, H4′, J = 5.2 Hz, J = 3.7 Hz); 6.96 (d, 1H, H4, J = 5.2 Hz); 3.81 (t, 2H, Halk, J = 6.9 Hz); 3.00 (t, 2H, Halk, J = 6.9 Hz). 13C NMR (50 MHz, CDCl3) δ, ppm: 135.7; 135.4; 132.3; 130.1; 127.6; 126.6; 125.8; 124.4; 62.8; 32.4. HRMS (MALDI–TOF) m/z: calculated for C10H10OS2 (+Na) = 233.0065; found (+Na) = 233.0059. Anal. calc. for C10H10OS2 : C 57.11, H 4.79; found: C 56.95, H 4.99.
3.12 Synthesis of (12a) and (12b)
Compounds 12a and 12b are light sensitive, usual precautions must be taken. To a solution of 329 mg (1.6 mmol) of 10 in 5 ml of dried THF was added dropwise, at 0 °C, 980 μl (1.6 mmol) of n-BuLi (1.6 M in hexane) or 61.1 mg of NaH (60% in mineral oil). The mixture was stirred for 10 min at 0 °C. A solution of 0.5 equiv of arenetricarbonylchromium 11a [41] or 11b [34] in THF was added to the reaction at 0 °C. The reaction was stirred at 0 °C for 10 min and at room temperature for four days. The mixture was poured into water, extracted with diethylether, dried over MgSO4 and evaporated under reduced pressure. The crude products can be also purified by silica-gel-column chromatography using a mixture of cyclohexane/dichloromethane: 50:50 for both products to yield 85% of 12a and 90% for 12b. Compound 12a: 1H NMR (200 MHz, CDCl3) δ, ppm: 7.33 (dd, 1H, H5′, J = 4.9 Hz, J = 1.5 Hz); 7.23 (d, 1H, H5, J = 4.9 Hz); 7.15 (dd, 1H, H3′, J = 3.4 Hz, J = 1.5 Hz); 7.07 (dd, 1H, H4′, J = 4.9 Hz, J = 3.4 Hz); 7.02 (d, 1H, H4, J = 4.9 Hz); 5.46 (dd, 1H, HArCr, J = 6.4 Hz, J = 1.5 Hz); 5.32 (td, 1H, HArCr, J = 6.4 Hz, J = 1.5 Hz); 5.01 (d, 1H, HArCr, J = 6.4 Hz); 4.89 (t, 1H, HArCr, J = 6.4 Hz); 4.04 (m, 2H, Halk); 3.26 (t, 2H, Halk, J = 6.9 Hz). 13C NMR (50 MHz, CDCl3) δ, ppm: 233.8; 140.5; 135.3; 134.0; 132.6; 130.1; 127.7; 126.8; 126.0; 124.6; 98.5; 97.0; 92.8; 86.6; 68.8; 28.7; 16.3. UV/visible (CHCl3): λ (nm) = 300; ϵ = 13 082 cm–1 mol–1 l. IR (KBr): 1867, 1954 cm–1 (νCO). Compound 12b: 1H NMR (200 MHz, CDCl3) δ, ppm: 7.33 (dd, 1H, H5′, J = 5.4 Hz, J = 0.98 Hz); 7.22 (d, 1H, H5, J = 5.4 Hz); 7.14 (dd, 1H, H3′, J = 3.4 Hz, J = 0.98 Hz); 7.07 (dd, 1H, H4′, J = 5.4 Hz, J = 3.4 Hz); 7.00 (d, 1H, H4, J = 5.4 Hz); 5.55 (t, 1H, HArCr, J = 6.9 Hz); 5.13 (t, 1H, HArCr, J = 2 Hz); 4.78 (td, 2H, HArCr, J = 6.9 Hz, J = 2 Hz); 4.05 (t, 2H, Halk, J = 6.9 Hz); 3.71 (s, 3H, CH3); 3.23 (t, 2H, Halk, J = 6.9 Hz). 13C NMR (50 MHz, CDCl3) δ, ppm: 233.6; 143.9; 143.1; 133.7; 130.0; 127.7; 126.7; 126.0; 124.6; 93.2; 73.4; 73.0; 69.7; 68.6; 55.8; 28.7. UV/Visible (CHCl3): λ (nm) = 302; ϵ = 14325 cm–1 mol–1 l. IR (KBr): 1870, 1952 cm–1(νCO).
Acknowledgements
The authors would like to thank the CNRS and the EU Training and Mobility of Researchers Program: ‘Organometallic Dipole with NLO Properties′ contract # ERBFMRXCT-CT98-0166 and COST D14 for financial support. We thank Prof. C. Rolando and Dr G. Ricart (UST Lille, France) for HRMS analyses.