

1 Introduction
For several years now, we have been investigating dinuclear organoiron complexes featuring various carbon-rich central spacers and possessing the electro-active terminal iron site (η5-C5Me5)Fe(η2-dppe), as depicted in Fig. 1 [1]. The detailed synthesis of most of these compounds has already been presented elsewhere [2–9]. These organometallics possess promising potential for the development of new nanoscopic devices in molecular electronics and especially for the construction of molecular wires. The one-electron oxidized complexes present a mixed-valence character and, as monoradicals, they are paramagnetic. The understanding of their magnetic properties is quite easy [1, 10, 11]. In particular, their magnetic susceptibility obeys the Curie Law. Early investigations on the diradical dication complexes deriving from the two-electron oxidation of the neutral species have revealed the paramagnetic character of these species. These symmetric dicationic diradicals, bridged by sp2- or sp-carbon linkers, possess two metal sites carrying each an unpaired electron in nearly degenerated singly occupied molecular orbitals. If there is a sizeable interaction between the unpaired electrons, two different spin configurations are expected, with different energy levels. The triplet configuration presents a magnetic moment and threefold degeneracy, whereas the singlet configuration is diamagnetic and non-degenerated. Depending on the ground state (triplet or singlet), the electronic spins will line up either in the same or in the opposite directions and are respectively said to interact in a ferromagnetic or antiferromagnetic fashion (Fig. 1). In addition, the singlet state can be stabilized by electronic correlation with closed-shell configurations. As a result, geometrical changes are generally associated with spin transitions and the overall geometry is usually driven toward a more-bonding arrangement of the atoms in the singlet state relative to the triplet state. However, in spite of its closed shell Lewis representation, the singlet ground state does not exactly correspond to a cumulene structure, but has also an open-shell (diradical) character. Whatever its correct structure, the singlet state is generally represented by a cumulene structure, but it can be more accurately described by the two Lewis structures A and B (Fig. 1) [10].
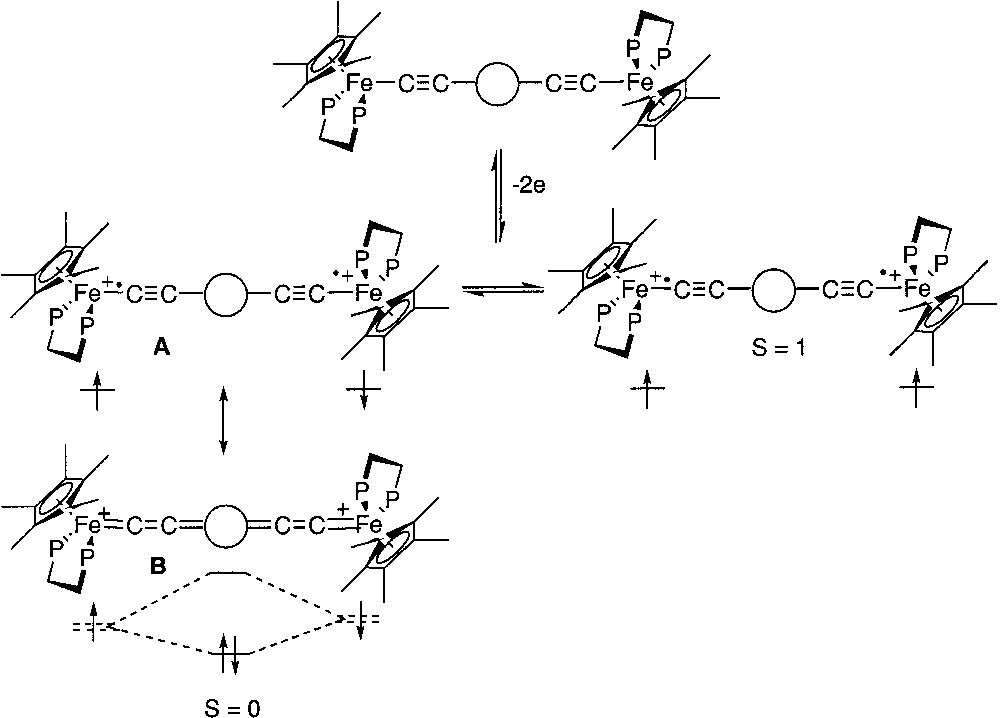
Dinuclear organoiron complexes featuring various carbon-rich central spacers and possessing the electro-active terminal iron site (η5-C5Me5)Fe(η2-dppe).
Among the physical methods that have been used to characterize the spin states, Mössbauer spectroscopy has proven to be a very efficient tool. Indeed, the quadrupole splitting (QS) of the 57Fe Mössbauer spectrum is closely related to the oxidation states of the metal nucleus. In the dicationic complexes depicted in Fig. 1, the open-shell configurations are closely related to iron(III) alkynyl structures, whereas the closed-shell configuration presents an iron(II) cumulene-like structure. It has been established on mononuclear model compounds that the QS values for these two types of structures range between 0.89 and 0.95 for the iron(III) derivatives and between 1.0 and 1.4 for the second group of compounds [12]. Curiously, the Mössbauer spectra recorded at 80 K for the binuclear dications represented in Fig. 1 exhibit a unique doublet. Since the two spins isomers are in equilibrium at this temperature, as shown from magnetic susceptibility measurements, this implies that they interconvert in the solid state at a fast regime with respect to the Mössbauer timescale (10–7 s). In the particular case of the diradical with the 1,4-dimethoxy-butadiene-1,4-diyl bridge (Fig. 2), the Mössbauer spectroscopy allows the simultaneous observation of two doublets. Their spectroscopic signatures (QS) remain clearly distinct between 4.5 and 293 K, and the relative amount of the spin isomers changes reversibly with the temperature, evidencing the thermal equilibrium between both species [13]. Coalescence was not approached, meaning that the exchange lifetime must be far above 10–6 s. Moreover, the variable temperature ratio of both isomers computed from the Mössbauer doublets surface areas allows the determination of singlet/triplet energy gap (ΔEST = –27 cm–1). This value is in good agreement with the value computed from the magnetic susceptibility measurements. In addition, the isomeric shift (δ) and quadrupole splitting parameters are suggestive of a Fe(II)=C bonding for the singlet state (δ = 0.23 mm s–1 vs Fe, QS = 1.06 mm s–1) and a Fe(III)–C for the triplet state (δ = 0.48 mm s–1 vs Fe, QS = 0.83 mm s–1).
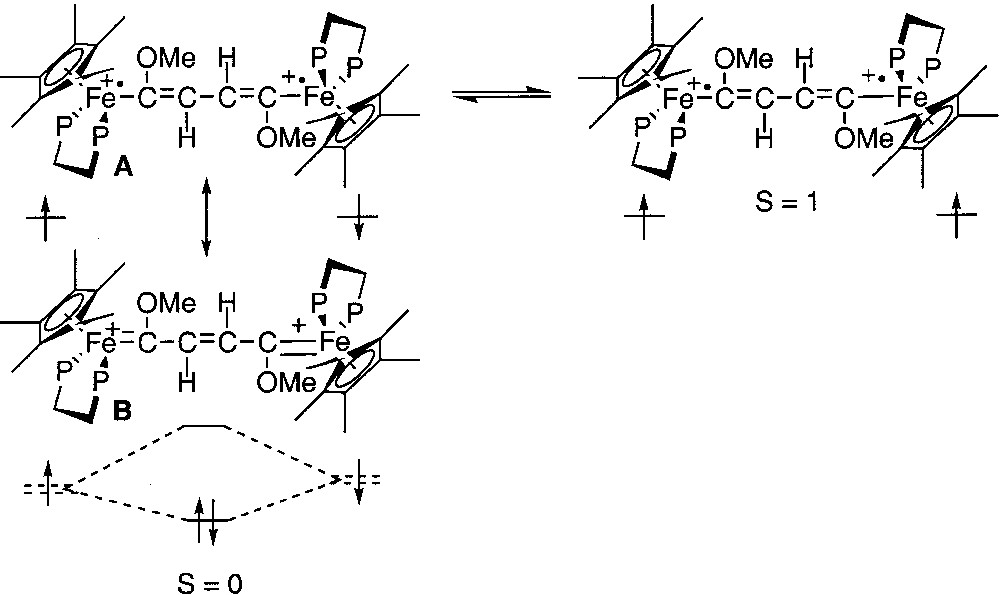
Diradical with the 1,4-dimethoxy-butadiene-1,4-diyl bridge.
Concomitantly to our activity on binuclear systems represented in Figs. 1 and 2, we have also an ongoing interest in the synthesis of mononuclear compounds to develop new spectroscopic means for investigating the electronic structure of the new compounds [12, 14, 15]. As a result of our previous research on model compounds in the (η5-C5Me5)Fe(η2-dppe) series, we found that the quadrupole splitting is diagnostic not only of the oxidation state of the iron nucleus, but also of the bond order of the iron–carbon bond between the metal and the end-bound carbon ligand. In particular, we have shown how mononuclear compounds having a cumulene structure (Fig. 3, structure B) can be differentiated from those having an alkynyl structure (Fig. 3, structures A and C). We have also noted that the QS value is significantly larger for compounds having a heteroatom on the α or γ carbon atoms of the carbon-rich ligand, suggesting that the QS value increases when the bond order decreases [12].
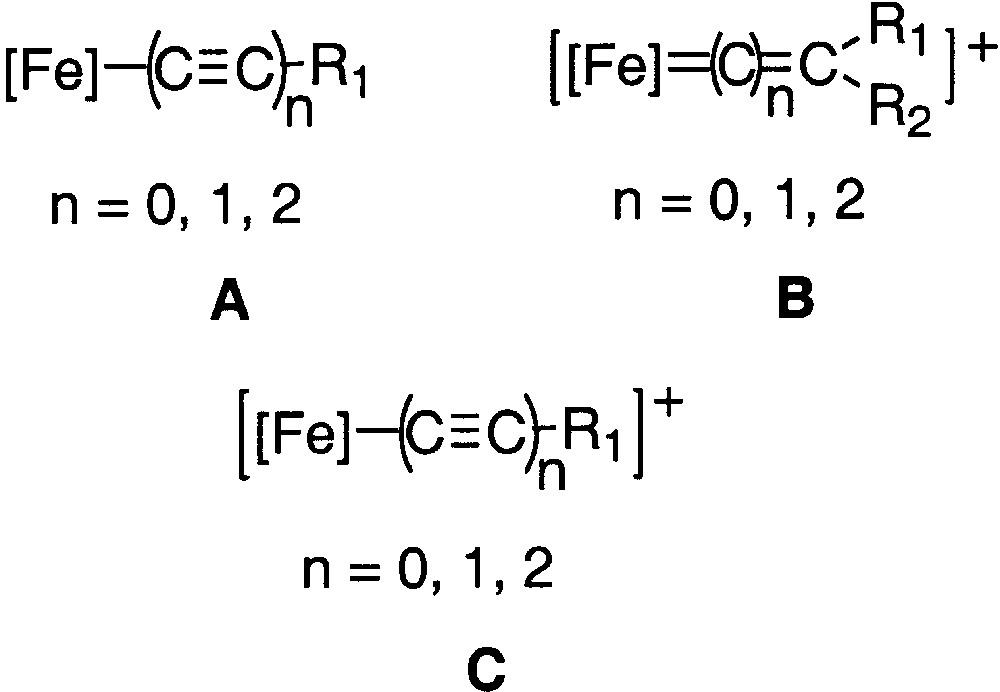
Mononuclear compounds with a cumulene structure (structure B) and with an alkynyl structure (structures A and C).
In order to obtain more in depth information on the nature of the Fe=C bond in compounds possessing the (η5-C5Me5)(η2-dppe)Fe framework and a cumulene-like ligand, we have carefully studied the variation of the iron-carbon bond distances determined by X-ray diffraction with the QS value of the Mössbauer spectrum for a selection of mononuclear iron complexes bearing carbene, vinylidene and allenylidene ligands as shown in Fig. 4. For this purpose: (i) the Mössbauer data available in the literature for the compounds 4 [16], 8 [12], and 9 [17] were collected; (ii) we have determined the Mössbauer parameters for the known carbene derivatives 1, and 2a; (iii) have run the Mössbauer spectra for the vinylidene 3; (iv) the new allenylidene complexes 5a, 5b, 6, and 7 were prepared and their spectroscopic characterizations achieved including the determination of the Mössbauer parameters; (v) single crystals suitable for X-ray diffraction analyses were grown for 3, 4, and 5a; (vi) in addition, Mössbauer spectra were run for the complexes 2b and 5b to investigate the possible effect of the counter-anions.
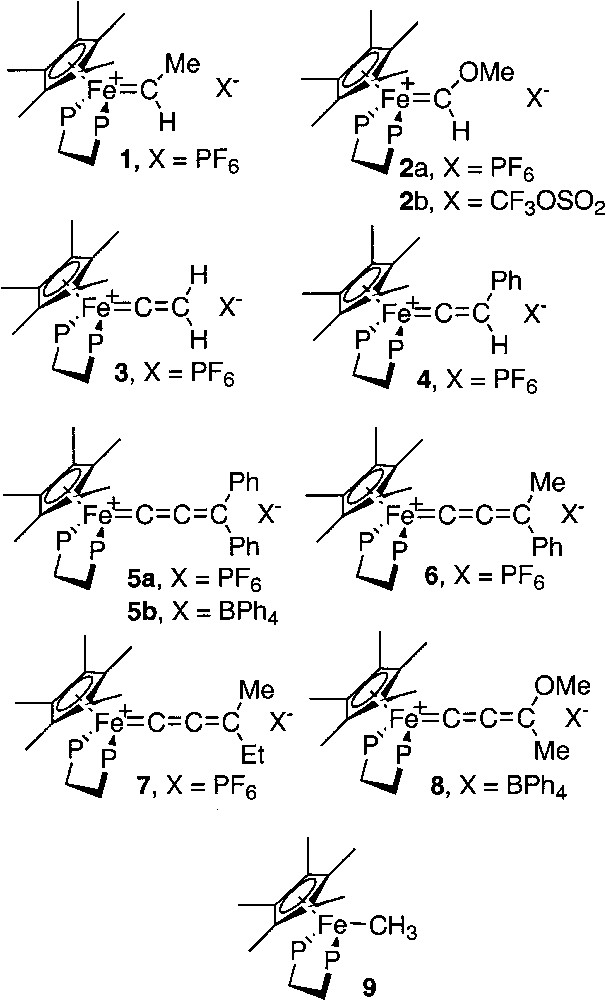
Mononuclear iron complexes bearing carbene, vinylidene and allenylidene ligands.
2 Results and discussion
2.1 Synthesis of the allenylidene complexes 5–7
Very few iron allenylidene complexes were reported in the [(η5-C5R5)(η2-dppe)Fe(=C=C=CR1R2)][X] series. To our knowledge, the preparation of [(η5-C5H5)(η2-dppe)Fe(=C=C=CPh2)][BF4] by photo-activation upon irradiation at 280 nm of [(η5-C5H5)(η2-dppe)Fe(CO)][BF4] in the presence of the propargylic alcohol H-C≡C–C(Ph)2OH constitutes a very rare example for such a complex [18]. To access to our synthetic targets, we followed the classical thermal procedure. This efficient method was first employed for the preparation of a ruthenium complex [19], but has found widespread application with other transition elements [20].
Complex (η5-C5Me5)(η2-dppe)FeCl (10) reacts with 1 equiv of 1,1-diphenyl-prop-2-yn-1-ol in methanol at 20 °C in the presence of KPF6 for 12 h to give the purple allenylidene complex [(η5-C5Me5)(η2-dppe)Fe(=C=C=C(Ph)Ph)][PF6] (5a, 95%). The analytically pure complex was characterized by IR and NMR. The IR spectrum of 5a shows typical νC=C=C and ν(PF6) at 1896 and 839 cm–1, respectively. The 31P{1H} NMR of 5a in CDCl3 displays a singlet for the dppe ligand and a septuplet characteristic of the PF6 anion. The allenylidene derivatives are often formed from their hydroxyvinylidene precursors by spontaneous loss of H2O, but sometimes this step requires assistance: for example, an acidic catalysis (Fig. 5). A salt effect can also facilitate the dehydration step. Indeed, in order to prepare the diphenylallenylidene iron complex as a tretraphenyl borate salt, we worked in the same conditions, replacing KPF6 by NaBPh4. Surprisingly, 5b was isolated as a mixture with the hydroxyvinylidene [(η5-C5Me5)(η2-dppe)Fe(=C=C(H)C(OH)(Ph)2)][BPh4] (14b) in the 1:1 ratio. The vinylidene intermediate was identified in the mixture by its IR characteristic vibration modes νC=C and νOH at 1616 and 3560 cm–1, respectively. The 1H NMR spectrum of complex 14b shows characteristic signals at δ 1.49 (s) and 4.82 (t, JHP = 5 Hz), corresponding to the resonances of the protons of the C5Me5 ligand and the β carbon of the vinylidene fragment. A resonance at 88.6 ppm was also observed in the 31P NMR spectrum. This signal presents the same intensity than that of 5b, allowing the determination of the ratio 5b/14b in the crude product. The salt effect on the dehydration step could be explained by the ability of the hexafluorophosphate to give hydrogen bonds with acidic protons. For example, a hydrogen bond was recently observed by X-ray between a PF6– anion and an acidic hydrogen atom in an organometallic compound [21]. In the case of the mixture of 5b and 14b, treatment of the crude product with Amberlyst 15 in methanol provides the allenylidene 5b as a pure sample. The preparation of 5b was also carried out in the presence of Amberlyst 15. In this case, the vinylidene intermediate was not observed and 5b was directly obtained in 91% yield. The allenylidene complexes 6 and 7 were prepared according to the same procedure and they were isolated as purple powders in 90% yield. Subsequent crystallization gave analytically pure compounds which were characterized by IR, 1H-, 13C-, 31P-NMR spectroscopies.
Formation of allenylidene derivatives from their hydroxyvinylidene precursors by spontaneous loss of H2O with the assistance of an acidic catalysis.
2.2 Crystal structures of 3, 4·C4H10O, and 5a·C4H10O
The crystal structures of 3, 4·C4H10O and 5a·C4H10O were determined as outlined in Table 1 and the ‘Experimental’ section. The molecular structures are shown in Figs. 6–8. Key distances and angles are given in Table 2. General features, such as the formally octahedral geometry at the iron centres, accord with past structures in this series [1, 11]. The Fe=C bond distances in 3 (1.763(7) Å) and 4·C4H10O (1.760(5) Å) are very similar and consistent with a bond order of about two. However, it can be observed that the Fe=C bond length in 3 and 4·C4H10O are shorter than the Fe=C bond in the related iron carbene 1 (1.787(9) Å) [11] or in the allenylidene 5a·C4H10O (1.781(9) Å). It is also noteworthy that theoretical calculations have shown that the metal–carbon bond in metallavinylidene derivatives possesses a partial triple bond character [22]. The Cα–Cβ bond distances in 3 (1.224(9) Å) and 4·C4H10O (1.295(5) Å) are relatively short for a C=C double bond [23, 24]. The Fe–Cα–Cβ angle slightly deviates from linearity as it was previously noted in the closely related vinylidene [(C5H5)(dppm)Fe(=C=C(Me)Ph)]I [24] and also in many other mononuclear complexes bearing a vinylidene ligand [23]. Moreover, the positions of the hydrogen atoms on Cβ were found by analysis of the X-ray data allowing the observation of a partial pyramidalization of the β carbon. In contrast, this carbon presents a planar geometry in the complex 4·C4H10O. Note that very few complexes of primary vinylidene with transition metal were X-ray characterized, and for the first row of the d-elements there is only one precedent [25].
Crystallographic data for 3, 4·C4H10O and 5a·C4H10O
| 3 | 4·C4H10O | 5a·C4H10O | |
| Molecular formula | C38H41F6FeP3 | C44H45F6FeP3·C4H10O | C51H49F6FeP3·C4H10O |
| Molecular weight | 760.51 | 910.68 | 998.78 |
| Crystal system | orthorhombic | monoclinic | triclinic |
| Space group | P212121 | P21/c | P–1 |
| Cell dimensions | |||
| a (Å) | 12.606(2) | 12.072(3) | 9.726(3) |
| b (Å) | 16.576(3) | 14.609(3) | 14.701(6) |
| c (Å) | 17.515(5) | 26.054(9) | 18.794(5) |
| α (deg) | 90.00 | 86.84(3) | |
| β (deg) | 90.00 | 102.41(3) | 79.32(3) |
| γ (deg) | 90.00 | 72.52(4) | |
| V (Å3) | 3660(2) | 4488(2) | 2519(2) |
| Z | 4 | 4 | 2 |
| Temperature (K) | 293 | 293 | 293 |
| dcalc (g cm–3) 293 K | 1.380 | 1.348 | 1.317 |
| Absorption coefficient (mm–1) | 0.595 | 0.503 | 0.455 |
| F(000) | 1576 | 1904 | 1044 |
| Crystal dimensions (mm) | 0.28 × 0.33 × 0.35 | 0.45 × 0.38 × 0.35 | 0.35 × 0.35 × 0.32 |
| Diffractometer | CAD4 Nonius | CAD4 Nonius | CAD4 Nonius |
| Radiation (Å) | Mo Ka (0.71073) | Mo Ka (0.71073) | Mo Kα (0.71073) |
| Data collection method | ω/2θ, 2θmax = 54° | ω/2θ, 2θmax = 54° | ω/2θ, 2θmax = 54° |
| tmax/measure (s) | 60 | 60 | 60 |
| range/indices (h,k,l) | 0, 16; 0, 21; 0, 22 | 0, 15; 0, 18; –33, 33 | 0,12; –18, 18; –23, 23 |
| θ range | 1.69 to 26.99 | 1.61 to 26.97 | 1.10 to 26.97 |
| Reflections measured | 4471 | 10 245 | 11 500 |
| Independent reflections | 3048 | 9778 | 10 964 |
| Obsd data, I > 2 θ(I) | 2810 | 5612 | 5500 |
| Number of variables | 543 | 528 | 571 |
| Final R | 0.0503 | 0.0513 | 0.0748 |
| R indices (all data) | 0.0859 | 0.696 | 0.1824 |
| Rw | 0.047 | 0.194 | 0.203 |
| GOF | 0.979 | 1.042 | 1.021 |
| Largest diffraction peak and hole (e Å–3) | residual Δρ < 0.38 | residual Δρ ≤ 0.9 | residual Δρ ≤ 1.21 |
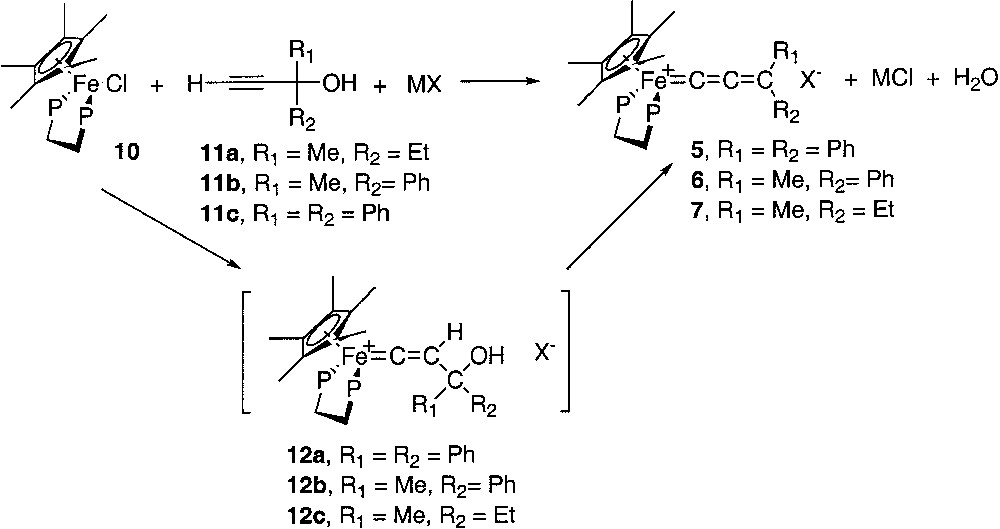
Molecular structure of [(η5-C5Me5)(η2-dppe)-Fe(=C=CH2)][PF6] (3). Non-hydrogen atoms are represented by 50% probability thermal ellipsoids.
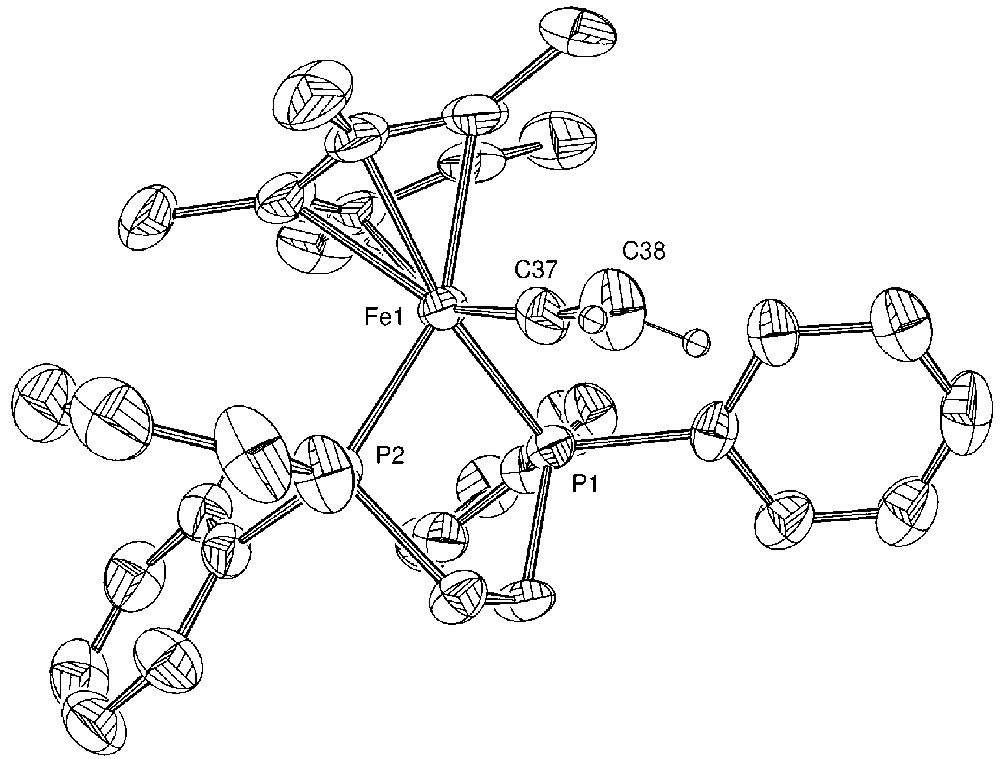
Molecular structure of [(η5-C5Me5)(η2-dppe)-Fe(=C=C(Ph)H)][PF6] (4). Non-hydrogen atoms are represented by 50% probability thermal ellipsoids.
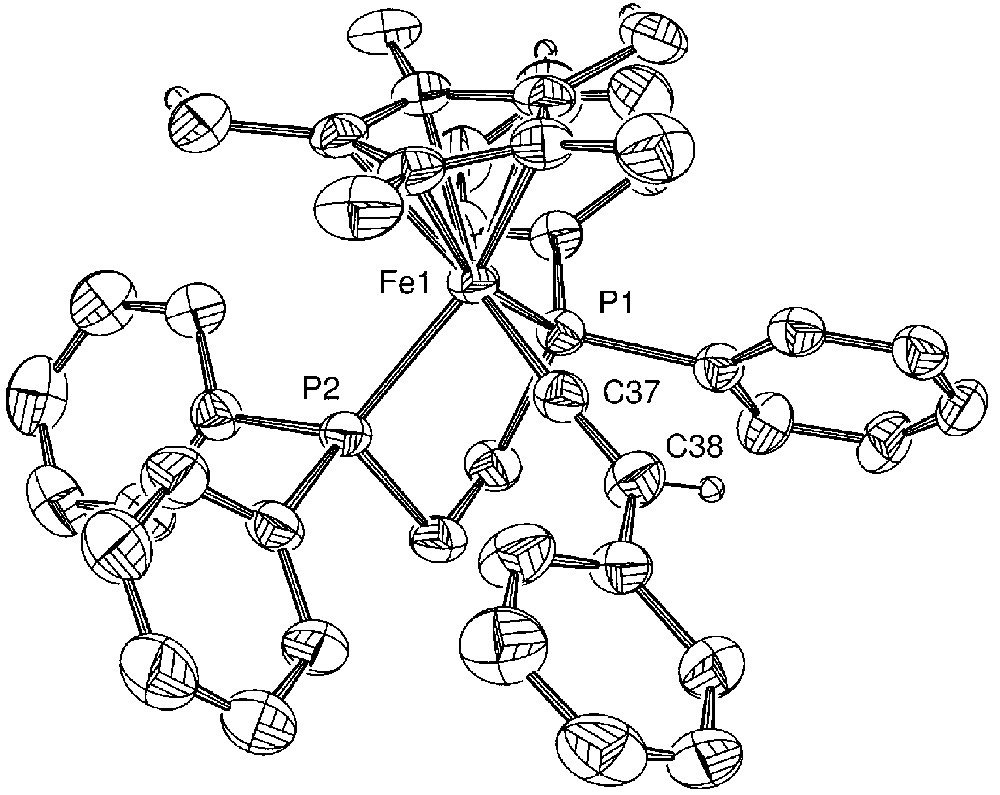
Molecular structure of [(η[(η5-C5Me5)(η2-dppe)-Fe(=C=C=C(Ph)Ph)][PF6] (5a). Non-hydrogen atoms are represented by 50% probability thermal ellipsoids.
Key distances (Å) and angles (deg) for 3 4·C4H10O, and 5a·C4H10O
| 3 | 4·C4H10O | 5a·C4H10O | |
| Fe1–P1 | 2.2219(15) | 2.2482(14) | 2.2344(18) |
| Fe1–P2 | 2.2185(16) | 2.2319(14) | 2.2186(16) |
| Fe1–C37 | 1.763(7) | 1.760(5) | 1.785(5) |
| C37–C38 | 1.224(9) | 1.295(5) | 1.257(7) |
| C38–C39 | 1.474(7) | 1.361(7) | |
| C39–C40 | 1.479(8) | ||
| C39–C46 | 1.470(7) | ||
| Fe1–C5Me5 (centroid) | 1.767 | 1.796 | 1.766 |
| P1–Fe1–P2 | 84.85(6) | 84.83(5) | 85.55(7) |
| P1–Fe1–C37 | 91.5(2) | 90.33(15) | 88.24(15) |
| P2–Fe1–C37 | 86.29(19) | 84.89(15) | 85.59(15) |
| Fe1–C37–C38 | 173.8(6) | 175.9(4) | 177.4(4) |
| C37–C38–H38A | 131.9 | 113.8 | |
| C37–C38–H38B | 129.2 | ||
| C37–C38–C39 | 132.3(5) | 177.6(5) | |
| C38–C39–C40 | 119.9(5) | ||
| C38–C39–C46 | 120.7(7) | ||
| C40–C39–C46 | 120.7(5) |
Although calculations have predicted that the plane of the vinylidene ligand should be perpendicular to the molecular plane in complexes of the type M(CCR2)(L2)(C5R5) [26], the barrier to rotation was computed to be only ca 15 kJ mol–1, so that this preference is often overridden by steric or packing effects [23]. This is the case in complex 3, where the values of the torsion angles defined by Cp*cent–Fe–C38–HA and Cp*cent–Fe–C38–HB are 91.1 and 125.6°, respectively. A similar behaviour is also noted in the case of complex 4·C4H10O, for which the torsion angles Cp*cent–Fe–C38–C39 and Cp*cent–Fe–C38–H are equal to 74.3 and 104.5°, respectively.
Structural parameters have been reported for a large number of mononuclear allenylidene complexes with many transition metals [20, 27]; however, only one precedent is known in the iron series [28]. As mentioned above, the Fe=C bond distance in the allenylidene 5a·C4H10O is 0.02 Å longer than in the vinylidene 3, but it compares very well with the value determined for the corresponding alkylidene derivative 1 [11]. As usual, in the three-carbon chain, the Cα–Cβ distances (1.257(7) Å) is shorter than the Cβ–Cγ distance (1.361(7) Å) [20]. These observed distances are not far from the values reported for similar diphenyl allenylidene complexes of ruthenium and osmium and indicate a contribution of the canonical form [M]-(C≡CC+Ph2) [20, 29]. Such an electronic contribution is also supported by EHMO calculations, which show that there is an alternation of electron density along unsaturated carbon chain. These studies indicate that Cα and odd-numbered carbon atoms are electrophilic centres, while Cβ and even-numbered atoms are electron rich [30].
The C3 fragment is nearly linear, the angles at Cα and Cβ being close to 180°. The plane defined by the carbon atom and the two ipso carbon atoms of the phenyl rings is almost perpendicular to the Cp* plane, since the torsion angles Cp*cent–Fe–C39–C40 and Cp*cent–Fe–C39–C46 are equal to 6.4 and 170.5°, respectively. The weak deviation from the orthogonal orientation theoretically expected for these two planes was also noted in several cases [20] and can be explained by the small barrier to rotation for allenylidene ligands, which was computed to be 11.3 kJ mol–1 for [(C5H5)(CO)2Fe (=C=C=CH2)]+ [22, 26].
2.3 Investigation of the iron–carbon bonding by Mössbauer spectroscopy
In the carbon-chain ligands (L =:C(=C)nRR’), the factors governing the metal–ligand interaction are electronic in character, as it was clearly established by studies on the relative orientation of the CR2 plane with respect to the symmetry plane of the CpML2 fragment and the determination of the rotation barrier for the unsaturated carbon fragment [22, 26]. The important orbitals in the description of the iron–carbon bonding are the empty π orbital, and the lone pair on the carbon fragment, which is invariably the HOMO. The characteristic feature of these ligands is the presence of one (carbene) or more π bonds between the metal and the terminal carbon, which presents a carbene character. The strength of the Fe–C π bond is the resultant of the balance between attractive and repulsive interactions that vary with the number of carbon atoms and the nature of the terminal R groups [22]. The usual spectroscopic properties like IR, 1H and 13C NMR, or UV–Visible do not provide clear information on the metal-carbon bonding. The more reliable data on the metal carbon-rich ligand interaction have been obtained by theoretical calculations and X-ray determination [20].
Mössbauer spectroscopy can shed some light on the strength of the π-bonding to cumulenylidene ligands. Indeed, the isomeric shift of the spectrum (δ) provides a rough indication of the electronic density at the iron nucleus and the width of the quadrupole doublet (QS) depends on several parameters including the anisotropic electron distribution in the valence shell of the Mössbauer atom [31]. Therefore, one can expect that the variation of the QS value mainly depends on the Fe=C bond distance. The zero-field Mössbauer spectra of the complexes 1–8 were run at 80 K and the characteristic parameters were computed. Careful examination of the data collected in Table 3 provides interesting information: in this homogeneous series of complexes, the δ and QS parameters roughly increase according to the same sequence (3 < 4 < 5a < 7 < 6 < 5b < 1 < 2a). This observation suggests that the positive charge on the iron nucleus decreases with the Fe=C bond order or, in other words, with the increase of the distance between the iron and the α carbon atom. Such a result is fully consistent with the valence bond (VB) analysis of the electronic structure of metallacumulenylidenes, as depicted in Fig. 9. When the π acidity of the carbon-rich ligand decreases, the weight of the VB structure B become less negligible; concomitantly, the electronic density on the metal increases and the Fe=C bond order decreases. In addition, in the case of R groups that contain an heteroatom, the predominance of the VB structure C would occur, especially in the case of metallacumulene possessing an odd number of carbon atoms [20, 32, 33]. Interestingly, a significant linear correlation (R = 0.81) between the Mössbauer parameters δ and QS clearly shows the dependence of the electronic density on the iron centre and the Fe=C bond distance for these complexes (Fig. 10). This relationship indicates a trend, but must be considered with caution, even in a homogenous series. Indeed, the methoxyallenylidene 8 lies far above the correlation depicted in Fig. 10, suggesting that the Fe=C bond in this complex is much weaker than one can deduce from the electronic density on the iron nucleus.
Mössbauer parameters and Fe=C bond distances for the metallacumulenes 1–8
| Compound | X | νC=(C)x=C (Nujol, cm–1) | δ (mm s–1) | QS (mm s–1) | Γ (mm s–1) | % area | d(Fe–C) (Å)a | Reference |
| 1 | PF6 | 0.148 | 1.095 | 0.150 | 100 | 1.787(9) | [12] | |
| 2a | PF6 | 0.164 | 1.153 | 0.117 | 100 | 1.82(2) | [12] | |
| 2b | CH3OSO2 | 0.216 | 1.223 | 0.131 | 100 | [12] | ||
| 3 | PF6 | 1612 | 0.086 | 1.032 | 0. | 100 | 1.762(8) | this work |
| 4 | PF6 | 1613 | 0.126 | 1.096 | 0.137 | 100 | 1.760(5) | this work |
| 5a | PF6 | 1896 | 0.139 | 1.077 | 0. | 100 | 1.781(9) | this work |
| 5b | BPh4 | 1888 | 0.158 | 1.135 | 0.146 | 100 | this work | |
| 6 | PF6 | 1913 | 0.145 | 1.129 | 0.143 | 100 | this work | |
| 7 | PF6 | 1931 | 0.141 | 1.123 | 0.143 | 100 | this work | |
| 8 | BPh4 | 1947, 1938 | 0.160 | 1.451 | 0.171 | 100 | [12] | |
| 9 | — | — | 0.15 | 1.95 | — | 100 | 2.075b | [17] |
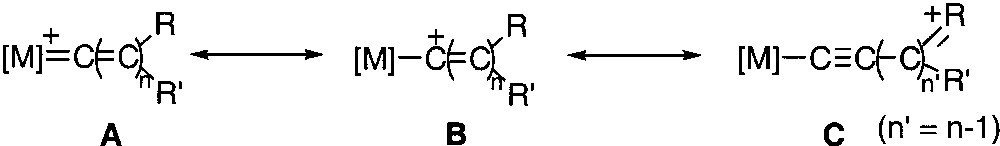
Electronic structure of metallacumulenylidenes.
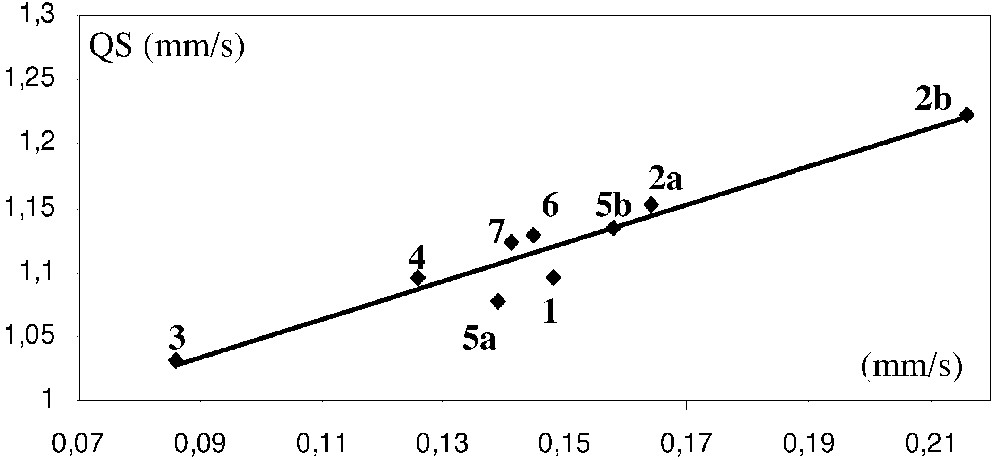
Plot of the Mössbauer quadrupole splitting (QS, mm s–1) of [(η5-C5Me5)(η2-dppe)Fe(=C(=C)nRR’)][X] complexes (1–7) vs the isomeric shift (δ, mm s–1 vs Fe).
An alternative means to investigate the effects of the structural variation of the ligand L on the metal–carbon bonding in this series of iron cumulenylidene derivatives is to correlate the QS values determined by Mössbauer spectroscopy with the Fe=C bond distances obtained by X-ray analyses. Fig. 11 displays a good linear dependence of these two terms (R = 0.99). However, considering that X-ray data are available for only the five complexes 1, 2a, 3, 4, and 5a, this observation has to be taken with caution. Nevertheless, it seems that for homogeneous series of metallacumulenes, the variation of the QS terms provides an indication on the strength of the π-bonding between the metal and the ligand. It is noteworthy that, in the limit of the accuracy of the X-ray determination, only 4 deviates from this linear correlation. Such a behaviour, which results from a shorter Fe=C bond distance that can be predicted from the QS value, could be related to the partial triple bond character of the Fe=C bond in the vinylidene derivatives, as pointed out by theoretical calculations [22].
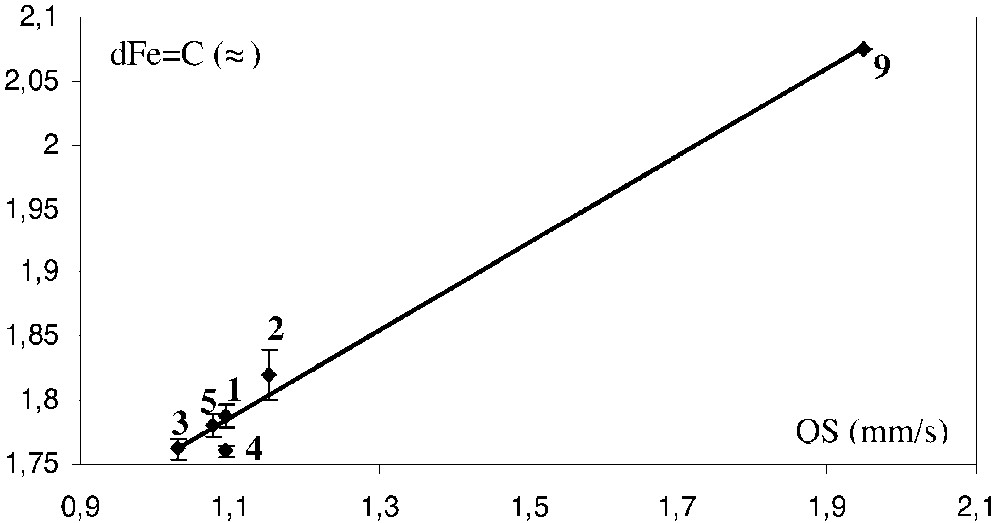
Plot of the iron-carbon bond distances (Å) of [(η5-C5Me5)(η2-dppe)Fe(=C(=C)nRR’)][X] complexes (1–5) and the [(η5-C5Me5)(η2-dppe)Fe(CH3)] derivative 9 vs the quadrupole splitting (QS, mm s–1).
Support on this conclusion can also be obtained by considering the iron alkyl complex (η5-C5Me5)(η2-dppe)Fe(CH3) (9) for which there is no π-bonding between the metal and the alkyl fragment. The QS value determined for this complex, which possesses a Fe-C bond with a pure σ-character, can be regarded as the limit value expected when the π-bonding interaction does not exist. The Fe–C bond distance was not determined by X-ray analysis for complex 9, but it was computed by DFT method (2.075 Å) [34]. The correlation in Fig. 11 was complemented by the data obtained for compound 9. Interestingly, these values confirm the trend observed for the cumulene derivatives and the fit remained unchanged (R = 0.99). The empiric dependence between the Fe=C distances and the QS value is given by equation (1).
| 1 |
The Mössbauer spectroscopy constitutes a convenient mean to obtain a powerful characterization of the double bond between the iron centre and the carbon-chain ligands. It is important to note that for the given (η5-C5Me5)(η2-dppe)Fe metal fragment, the Mössbauer parameters are very sensitive to the structure of the cumulene ligand L. However, the great sensitivity of the Mössbauer spectroscopy to the nature of the counter anion must be also emphasized. Indeed, comparison of the data obtained for the couples of compounds 2a/2b and 5a/5b clearly shows that the effect induced on the Mössbauer response of the iron nucleus by the replacement of the PF6– counter anion by CF3OSO2– or BPh4– can be much larger than the effect of the structural modifications on the cumulene fragment. Indeed, the quadrupole splitting (QS) results from the interaction between the nuclear quadrupole moment and the electric field gradient (EFG) at the nucleus. Distant ions which surround the Mössbauer atom clearly contribute to the total EFG, and consequently, anions with different volumes and symmetry could give rise to different QS values [31].
On the other hand, the relationship between the QS and δ parameters depicted in Fig. 10 shows that both the couples of complexes 2a, 2b and 5a, 5b fit very well with the other data of this empiric correlation. In addition, the absorption corresponding to the νC=C=C stretch is shifted to lower wavenumbers in 5b (1888 cm–1) relative to 5a (1896 cm–1), in accord with a weaker Fe=C bond in 5b than in 5a. The nature of the cation-anion interaction is not currently clear, but nevertheless these results suggest that the anion could contribute either to strengthen or to weaken the Fe=C double bonds in these cationic metallacumulenes. Examination of the X-ray crystal structures of 1, 2a, 3, 4, and 5a does not reveal any short distances between the fluorine atoms of the hexafluorophosphate anion and hydrogen atoms of the cationic moiety as recently evidenced in the organometallic compound [(C5H5)Fe(p-MeC6H4)–NHN=CH–C6H4–CH=CH–(C5H4)Fe(C5H5)]+PF6– [21]. In the absence of structural determination with anions different from the hexafluorophosphate, it is assumed that the influence of the anions is probably due to either Coulombic interactions or packing effects. According to the empiric relationship represented by equation (1), the replacement of the hexafluorophosphate by triflate or tetraphenyl borate in 2a or 5a could induce a lengthening of the Fe=C bond of ca 0.2 Å. In the absence of experimental structural data, this hypothesis still remains an open question.
3 Conclusions
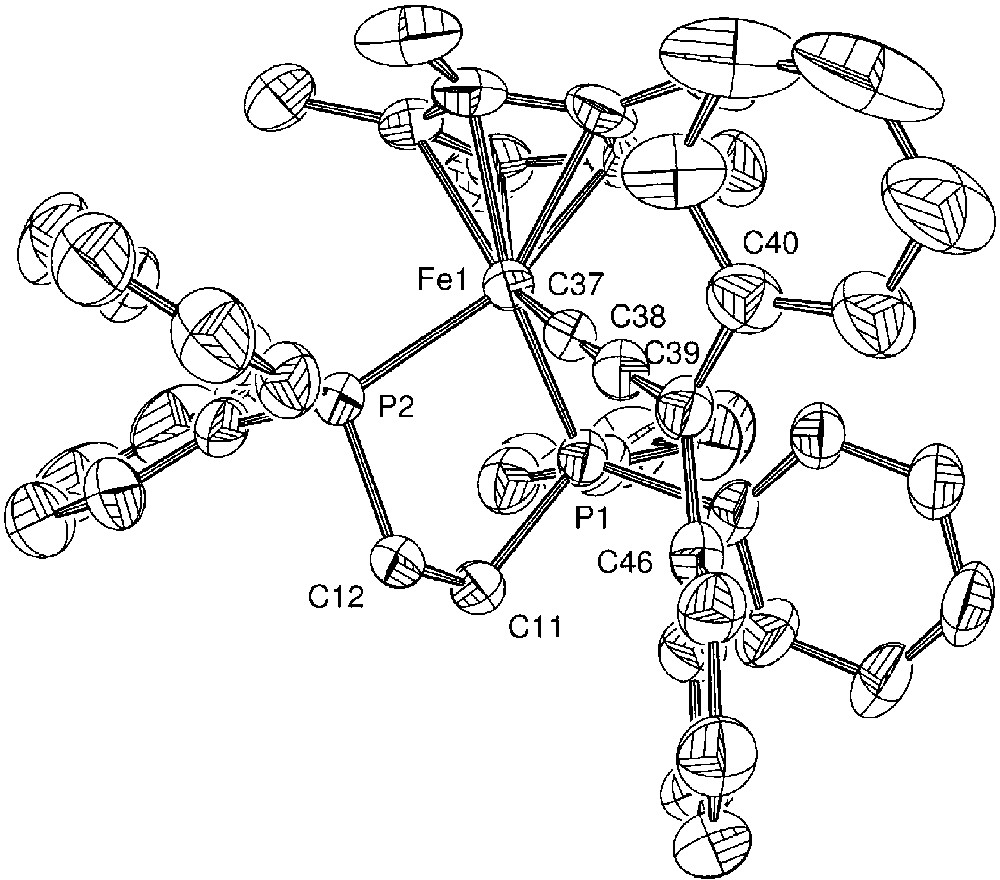
Mössbauer spectroscopy provides a better understanding of the Fe=C bonding in mononuclear metallacumulenylidenes. This spectroscopic mean is also helpful to investigate the bonding in the dinuclear dication complexes bridged by sp2- or sp-carbon spacers as shown in Figs. 1 and 2. In particular, in the case of the complex represented in Fig. 2, for which it was possible to observe two distinct Mössbauer doublets, it is of interest to compare the δ and QS parameters obtained for the singlet ground state (see introduction section) with the parameters characteristic for the mononuclear cumulene derivatives. The parameters of the singlet state are typical of Fe(II) derivatives, but they do not correlate with the data represented in Fig. 3, indicating that the Fe=C bonding in the binuclear complex is different from those of the mononuclear compounds depicted in Fig. 4. This should reflect the significant contribution of the open-shell structure A in the VB description of their electronic structure. Recently, we have prepared a new compound with an anthracene fragment in the carbon-rich bridge, which spans the two organoiron building blocks (17, Fig. 12).
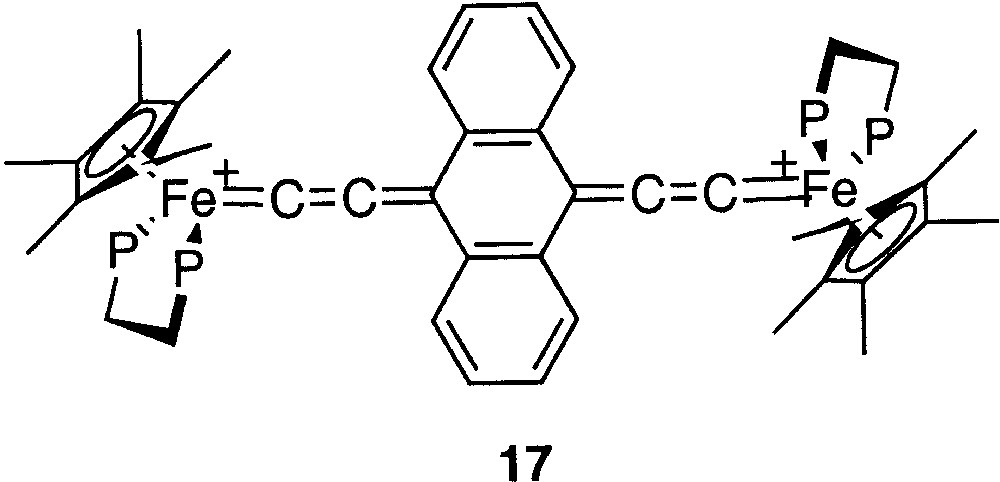
Compound 17, with an anthracene fragment in the carbon-rich bridge, which spans the two organoiron building blocks.
The Mössbauer parameters (δ = 0.171 mm s–1 vs Fe; QS = 1.120 mm s–1) fit very well with the correlations of Fig. 3. Moreover, the Fe=C carbon distance determined by X-ray (dFe=C = 1.819 Å) very slightly deviates from the correlation of Fig. 4 [35]. These data are in full agreement with the diamagnetic character of complex 17, determined by various means and support a very strong cumulene contribution for the electronic structure for the carbon-rich bridge containing the anthracene group.
4 Experimental section
4.1 General data
All manipulations were carried out under inert atmosphere. Solvents or reagents were used as follow: Et2O, THF, and n-pentane, distilled from Na/benzophenone; CH2Cl2, distilled from CaH2 and purged with argon; complexes [(η5-C5H5)2Fe+][PF6–] [36], and (η5-C5Me5)Fe(η2-dppe)Cl (10) [37] were prepared by previously published procedures. High-field NMR spectra experiments were performed on a multinuclear Bruker 300 MHz or 200 MHz instrument (AM300WB and 200DPX). Chemical shifts are given in parts per million relative to tetramethylsilane (TMS) for 1H- and 13C-NMR spectra, H3PO4 for 31P-NMR spectra. Transmittance–FTIR spectra were recorded using a Bruker IFS28 spectrometer (400–4000 cm–1). Mössbauer spectra were recorded with a 2.5 × 10–2 Ci (9.25 × 108 Bq) 57Co source using a symmetric triangular sweep mode [38]. LSI–MS analyses were performed at the ‘Centre régional de mesures physiques de l’Ouest’ (CRMPO, Rennes) on a high-resolution MS/MS ZabSpec TOF Micromass spectrometer (8 kV). Elemental analyses were performed at the Centre for Microanalyses of the CNRS at Lyon-Solaise, France.
4.2 [(η5-C5Me5)(η2-dppe)Fe(=C=C=C(Ph)Ph)][PF6] (5a)
In a Schlenk tube, the green complex 10 (0.300 g, 0.48 mmol), KPF6 (0.1 g, 0.54 mmol), methanol (30 ml) and 1,1-diphenylprop-2-yn-1-ol ( 0.110 g, 0.54 mmol) were introduced under argon. The mixture was stirred at 20 °C for 12 h. Evaporation of the solvent, extraction of the solid residue with methylene chloride, removal of the solvent and washing with n-pentane yielded the desired complex as a pure purple powder (0.420 g, 95%) after drying in vacuo. Crystals were grown by slow diffusion of diethyl ether in a dichloromethane solution of 5a. Anal. calcd for C51H49FeP3F6: C, 66.24; H, 5.34. Found: C, 66.75; H, 5.37. FT–IR (ν, KBr/Nujol, cm–1) 1896 (m, =C=C=C). NMR 1H (δ, CDCl3, 25 °C) 7.84–6.81 (30H, HAr/dppe); 3.01, 2.55 (2 m, 4H, CH2dppe); 1.46 (s, 15H, C5(CH3)5). NMR 31P (δ, CDCl3, 25 °C) 92.1 (s, dppe); –143 (sept, PF6). NMR 13C (δ, CDCl3, 25 °C) 285.0 (t, 2JCP = 37 Hz, Fe==C=C); 224.2 (s, Fe=C==C); 144.6 (s, Fe=C=C=); 135.1–128.3 (m, Ph); 101.8 (s, 5(CH3)5); 32.2, 31.3 (2m, –H2dppe); 10.5 (q, 1JCH = 127 Hz, C5(H3)5).
4.3 [(η5-C5Me5)(η5-dppe)Fe(=C=C=C(Ph)Ph)][BPh4] (5b)
In a Schlenk tube, the green complex (10) (0.500 g, 0.8 mmol) NaBPh4 (0.301 g, 0.88 mmol), Amberlyst 15 (0.500 g), methanol (30 ml) and 1,1-diphenylprop-2-yn-1-ol (0.183 g, 0.88 mmol) were introduced under argon. The mixture was stirred at 20 °C for 12 h. After evaporation of the solvent, MgSO4 was added before extraction of the solid residue with methylene chloride. Removal of the solvent and washing with n-pentane yielded the desired complex as a pure purple powder (0.800g, 91%). FT-IR (ν, KBr/Nujol, cm–1) 1888 (m, =C=C=C). NMR 1H (δ, CDCl3, 25 °C) 7.31–6.87 (50H, HAr/dppe); 2.89, 2.57 (2 m, 4H, CH2dppe); 1.42 (s, 15H, C5(CH3)5). NMR 31P (δ, CDCl3, 25 °C) 92.3 (s, dppe); –143 (sept, PF6). NMR 13C (δ, CDCl3, 25 °C) 284.8 (t, 2JCP = 37 Hz, Fe==C=C); 223.9 (s, Fe=C==C); 144.5 (s, Fe=C=C=); 166.1–163.2, 136.8–121.9 (m, Ph); 101.6 (s, 5(CH3)5); 32.1, 31.2 (2m, –H2dppe); 10.4 (q, 1JCH = 127 Hz, C5(H3)5).
4.4 [(η5-C5Me5)(η5-dppe)Fe[=C=C=C(Me)Ph][PF6] (6)
In a Schlenk tube, the green complex (10) (0.500 g, 0.8 mmol), KPF6 (0.162 g, 0.88 mmol), Amberlyst 15 (0.500 g), methanol (30 ml) and 1,1-diphenylprop-2-yn-1-ol (0.0 g, 0.88 mmol) were introduced under argon. The mixture was stirred at 20 °C for 12 h. After evaporation of the solvent, MgSO4 was added before extraction of the solid residue with methylene chloride. Removal of the solvent and washing with n-pentane yielded the desired complex as a pure purple powder (0.800 g, 91%). FT–IR (ν, KBr/Nujol, cm–1) 1913 (m, =C=C=C). NMR 1H (δ, CDCl3, 25 °C) 7.60–7.25 (25H, HAr/dppe); 3.09, 2.63 (2 m, 4H, CH2dppe); 1.70 (s, 3H, Me), 1.50 (s, 15H, C5(CH3)5). NMR 31P (δ, CDCl3, 25 °C) 92.1 (s, dppe); –143 (sept, PF6). NMR 13C (δ, CDCl3, 25 °C) 289.9 (t, 2JCP = 38 Hz, Fe==C=C); 218.2 (s, Fe=C==C); 143.2 (s, Fe=C=C=); 148.5–126.6 (m, Ph); 101.0 (s, 5(CH3)5); 32.2–31.4 (m, –H2dppe); 30.3 (q, 1JCH = 130 Hz, Me); 10.5 (q, 1JCH = 128 Hz, C5(H3)5).
4.5 [(η5-C5Me5)(η5-dppe)Fe(=C=C=C(Me)Et)][PF6] (7)
In a Schlenk tube, the green complex (10) (0.500 g, 0.8 mmol) NaBPh4 (0.301 g, 0.88 mmol), Amberlyst 15 (0.500 g), methanol (30 ml) and 3-methylpent-1-yn-3-ol (0.086 g, 0.88 mmol) were introduced under argon. The mixture was stirred at 20 °C for 12 h. After evaporation of the solvent, MgSO4 was added before extraction of the solid residue with methylene chloride. Removal of the solvent and washing with n-pentane yielded the desired complex as a pure purple powder (0.590 g, 91%). FT-IR (ν, KBr/Nujol, cm–1) 1931 (m, =C=C=C). NMR 1H (δ, CDCl3, 25 °C) 7.59–7.14 (20H, Hdppe); 3.01, 2.55 (2 m, 4H, CH2dppe); 1.79 (q, 3JHH = 7 Hz, 2H, C2–CH3); 1.47 (s, 3H, Me); 1.41 (s, 15H, C5(CH3)5); 0.86 (t, 3JHH = 7 Hz, 3H, CH2C3). NMR 31P (δ, CDCl3, 25 °C) 92.7 (s, dppe); –143 (sept, PF6). NMR 13C (δ, CDCl3, 25 °C) 301.9 (t, 2JCP = 37 Hz, Fe==C=C); 208.4 (s, Fe=C==C); 162.3 (s, Fe=C=C=); 135.0–128.6 (m, Ph); 100.0 (s, 5(CH3)5); 40.1 (t, 1JCH = 126 Hz, H2CH3); 32.8 (q, 1JCH = 126 Hz, CH3); 31.2 (2m, –H2dppe); 10.2 (q, 1JCH = 128 Hz, C5(H3)5). It was observed by 1H and 31P NMR spectroscopy that 7 slowly decomposes in (CD3)2CO, CD2Cl2 and CDCl3.
4.6 Crystallography
Data were collected on crystals of 3, 4·C4H10O and 5a·C4H10O as summarized in Table 1 [39, 40]. Cell constant and orientation matrix were obtained from a least-squares refinement using 25 high-θ reflections. After Lorentz and polarization corrections [41] and absorption corrections (φ scans), the structure was solved with SIR-97 [42], which revealed the non-hydrogen atoms and the solvate molecules. After anisotropic refinements, a Fourier difference map revealed many hydrogen atoms. Atomic scattering factors were taken from the literature [43]. ORTEP views were generated with PLATON-98 [44]. All calculations were performed on a Pentium NT Server computer.
4.7 Supplementary material
The supplementary material has been sent in electronic format to the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK as cif file No. CCDC 197018–97020, and can be obtained by contacting the CCDC.
Acknowledgements
We are grateful to A. Mari (‘Laboratoire de chimie de coordination du CNRS’, Toulouse, France) for Mössbauer measurements.