

1 Introduction
Molecular systems of defined architectures that respond to exogenous stimuli such as photons or electrons are of fundamental interest and are key design targets for future nanoscale devices. Coordination compounds stand out as advantageous functional components in such responsive systems due to their tremendous versatility in redox and photophysical features together with well-defined, yet diverse, coordination geometries. The incorporation of metal centers into materials provides, therefore, exceptional opportunities to control their electronic, magnetic and optical properties. Smart materials possessing novel physical and chemical properties that differ significantly from those accessible in ‘purely’ organic- or inorganic-based compounds can potentially be fabricated.
A productive approach for the incorporation of such useful chemical entities into materials must be versatile, modular and synthetically simple. Almost a decade ago, we embarked on a program directed at advancing the organic chemistry of coordination compounds, where functionalized complexes are viewed as synthetic building blocks. Our goal was to explore the prospects of a synthetic approach where the complexity of ‘inorganic’ complexes is increased via established ‘organic’ transformations. Instead of synthesizing intricate ligands, selective synthetic transformations are conducted on functionalized coordinatively saturated complexes, facilitating the exclusive formation of modified complexes. The purpose of this article is to describe the evolution of this program and highlight selected examples. Note that this mini-account is not meant to be comprehensive, but rather focused on developments in our own laboratories [1].
2 1,10-phenanthroline and its functionalization
Mono- and multimetallic compounds are invaluable for the study of energy and electron transfer processes [2], solar energy conversion [3], the construction of nanostructured materials [4], as well as the fabrication of molecular devices and sensors [5,6]. RuII complexes of polypyridine ligands play a central role in these explorations due to their chemical stability, and favorable redox and photophysical characteristics [7,8]. The most commonly used parent polypyridine ligands are 2,2′-bipyridine (bpy) 1, 2,2′:6,2′′-terpyridine (tpy) 2, and 1,10-phenanthroline (phen) 3, shown in Fig. 1. In spite of the numerous methods for synthesizing bpy and tpy ligands [9,10], we have favored the phen skeleton due to its advantageous characteristics, particularly its rigid structure holding the chelating nitrogens in juxtaposition [11]. A key step for our program was the strategic functionalization of phen along the long axis of the molecule (i.e., along the 3–8 positions). In collaboration with Siegel, we found out that treating the commercially available 1,10-phenanthroline monohydrochloride monohydrate with bromine in nitrobenzene (or bromobenzene) gives 3-bromo-1,10-phenanthroline 4 and 3,8-dibromo-1,10-phenanthroline 5 as the predominant products [12].
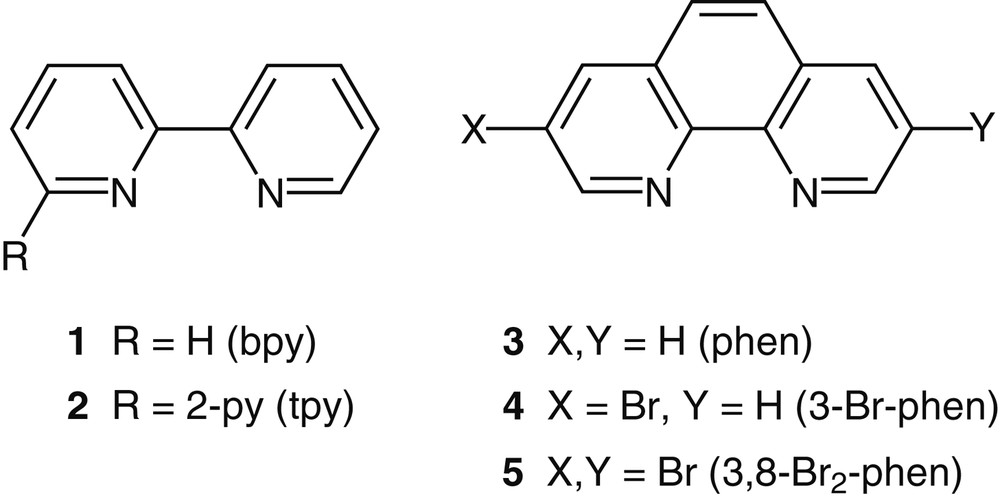
The parent polypyridine ligands: 2,2′-bipyridine 1, 2,2′:6,2′′-terpyridine 2, and 1,10-phenanthroline 3. Also shown are 3-bromo-1,10-phenanthroline 4 and 3,8-dibromo-1,10-phenanthroline 5.
3 Functionalized complexes and their reactivity
With the brominated phenanthrolines in hand, functionalized complexes such as 6–9 have been prepared and their synthetic chemistry explored (Fig. 2). Our early interest in conjugated metal containing systems has directed us to explore the utility of such functionalized building blocks in cross-coupling reactions. Such reactions have become extremely instrumental in constructing complex organic molecules [13], as well as oligomeric and polymeric materials [14,15]. The cross-coupling reactions of complex 6 with simple aromatic acetylenes were fast, clean and high yielding (Fig. 3) [16,17]. The high efficiency of these transformations was attributed to two main factors: (i) ‘protection’ of the phen nitrogens from coordinating the transition metal catalysts, and (ii) the electron deficiency of the RuII-coordinated 3-bromo-1,10-phenanthroline that facilitates the oxidative-addition of the catalytically-active Pd(0) species across the phen–Br bond. This high reactivity of these coordination compounds allowed us to construct multimetallic arrays in one step [16].
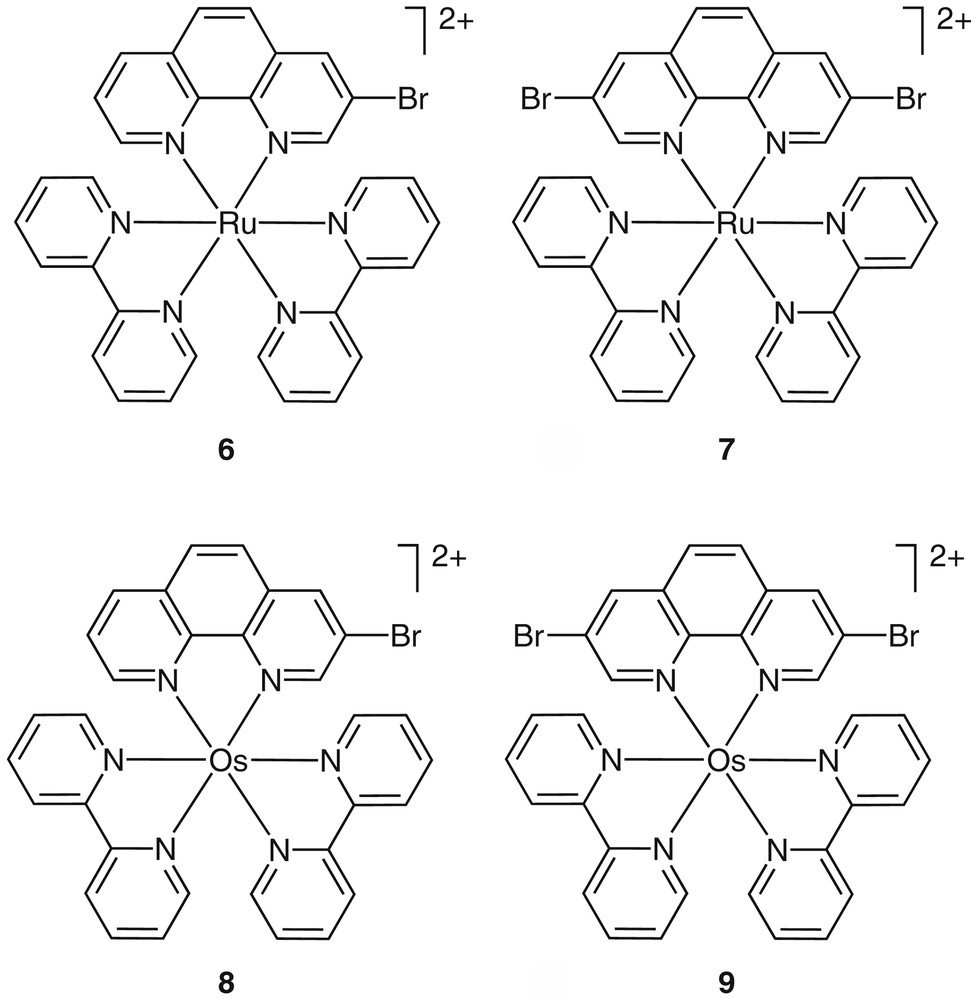
Representative examples for functionalized RuII and OsII coordination compounds. All complexes are typically isolated and used as their PF6– salts.
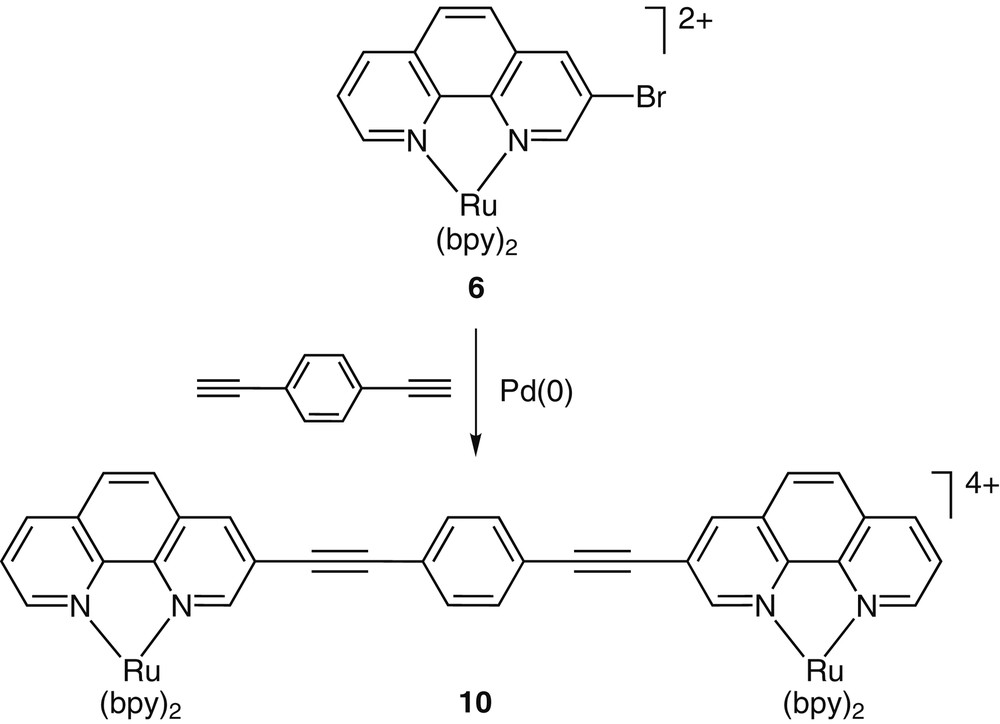
A Sonogashira cross-coupling reaction between the functionalized coordination compound 6 and a dialkyne provides the dinuclear complex 10 in high yield (86%).
The functionalized RuII building blocks can easily be prepared in their enantiomerically pure form [18]. This facilitates the synthesis of diastereomerically-pure multimetallic arrays (Fig. 4) [18]. Cross-coupling reactions performed on the periphery of these kinetically rather inert complexes do not cause any epimerization. Full control over the absolute configuration at each metal center can therefore be achieved. An example for a one-step synthesis of a meso dinuclear complex Δ,Λ–12 is depicted in Fig. 4 [18].
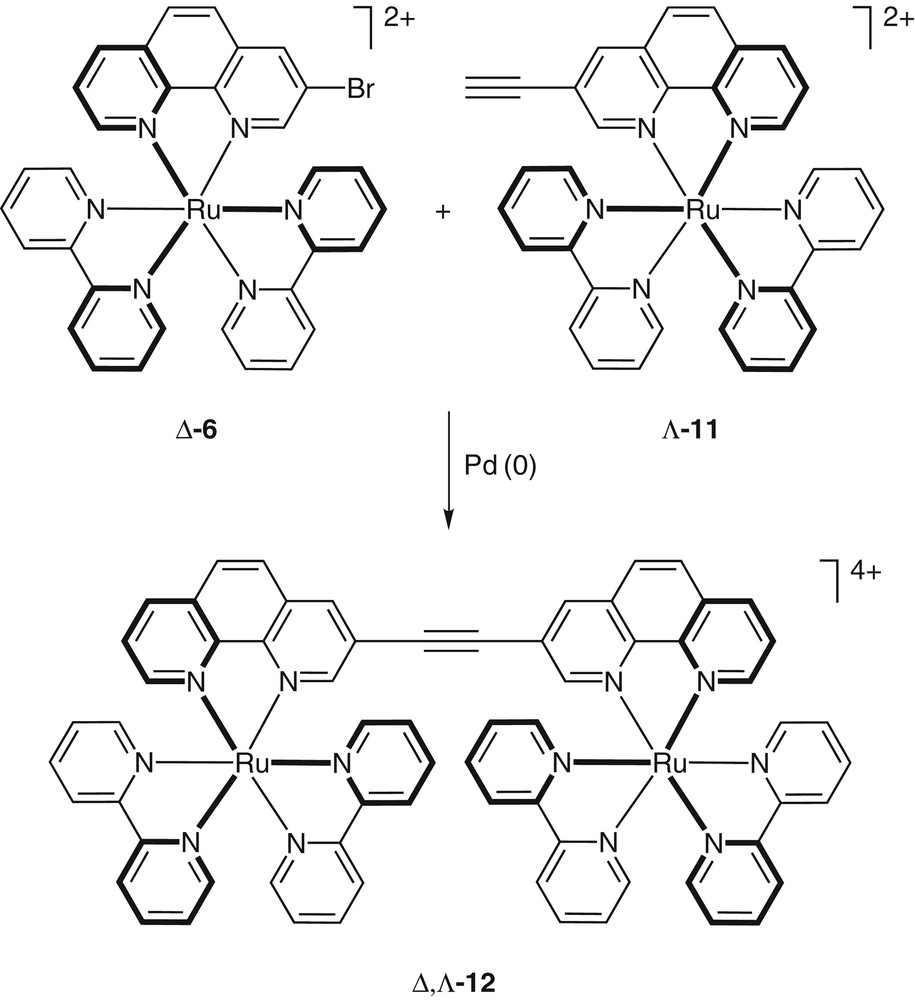
Enantiomerically pure coordination compounds as synthetic building blocks: Pd(0)-catalyzed cross-coupling reactions afford diastereomerically-pure dinuclear complexes in one step.
Our ‘coordination compounds as synthetic building blocks’ approach can also be applied to the synthesis of heterometallic arrays containing OsII and RuII [19]. Such molecules have extensively served as models for exploring electron and energy transfer processes [7,8]. Fig. 5 illustrates the single-step synthesis of a RuII–OsII diad 14. Controlling the structure and metal composition is achieved via the judicious selection of the appropriate building blocks. This approach facilitates the preparation of oligomeric heterometallic structures, such as RuII–OsII–RuII or OsII–RuII–OsII triads, which are practically inaccessible by alternative approaches [19].

Palladium-catalyzed cross-coupling reaction between functionalized RuII and OsII building blocks affords a hetero-dinuclear complex in one step.
The scope of reactions and suitable substrates is wider than may seem from the selected examples shown so far. Other cross-coupling transformations, in addition to the Sonogashira reaction, can be employed. For example, the brominated complexes (e.g., 6) are useful substrates for the Heck cross-coupling reaction [20]. It is also worth noting that numerous octahedral coordination compounds that possess other metal ions (e.g., RhIII), other functionalized ligands (e.g., 4-bromo- and 4-chloro-1,10-phenanthroline), different inert co-ligands (e.g., CO instead of bpy), as well as a number of reactive sites (as in [Ru(3-Br-phen)3]2+, for example), can be further derivatized using cross-coupling reactions. Similar cross-coupling reactions with the enantiomerically pure dibromo complex 7 and dialkynes yield conjugated stereochemically-defined metal-containing oligomers [21].
The successful application of metal-catalyzed cross coupling reactions to functionalized coordination compounds has prompted us to explore the scope of other transformations, in particular nucleophilic aromatic substitutions. The effect metal ions exert on substitution reactions of coordinated ligand has been of interest for decades [22]. Although intriguing observations have been made with organometallic species [23], less attention has been given to polypyridyl coordination compounds [24]. Early examples suggested that metal ion coordination could substantially alter the reactivity of 5-chloro-1,10-phenanthroline toward nucleophiles [25]. More recently, substitution reactions of RuII-coordinated 4,4′-dichloro-2,2′-bipyridines [26] and 4′-chloro-2,2′:6′,2′′-terpyridines have successfully been developed and utilized for extending the complexes into dendritic framework [27]. Whereas the 4-position of an uncoordinated or coordinated pyridine ring is inherently electrophilic, the 3-position is not [28]. We discovered, however, that substitution reactions with metal coordinated 3-bromo-1,10-phenanthroline (e.g., 6) were successful (Fig. 6), particularly with soft nucleophiles (e.g., thiolates) [29]. Other nucleophiles, such as alkoxide and phenoxide ions, worked reasonably well, while very hard nucleophiles, such as the fluoride ion, reacted only modestly [29]. In contrast to the effective reactions of the coordinated ligand, the free 3-bromo-1,10-phenanthroline was unsusceptible to similar nucleophiles under comparable conditions. We have therefore demonstrated that metal coordination indeed activates the meta position to the 1,10-phenanthroline’s nitrogen, a position previously assumed to be unreactive.
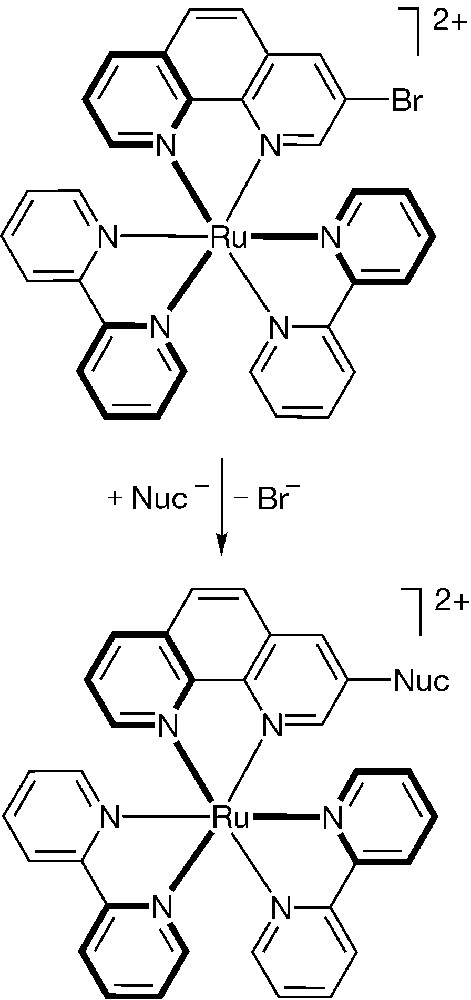
Functionalized coordination compounds can be substituted with various nucleophiles (M = Ru, Os; Nuc = RS, RO, ArO, Ar2P, F).
We have attributed the unprecedented reactivity of coordinated 3-substituted phen to the increased electrophilicity of the complexed ring upon metal coordination and the effective resonance stabilization of the anionic addition intermediate into the unsubstituted pyridine ring and its metal-nitrogen bond [29]. The latter effect is brought about by the conjugation of the two coordinated pyridine rings. The proposed mechanism shares many similar features with ‘traditional’ nucleophilic aromatic substitutions on electron-deficient arenes.
4 RuII complexes of large surface ligands
RuII–polypyridyl complexes are typically synthesized via a complexation reaction of the ligand of interest with an appropriate metal precursor. In contrast to the abundance of complexes that possess simple bpy, tpy and phen ligands, the repertoire of RuII complexes that contain large, extended surfaces is limited. The enormous potential of complexes that contain extended surfaces as nucleic acids intercalators, luminescent probes, as well as energy and electron donors and acceptors, have prompted us to develop new methodologies for their preparation.
We have recently reported an elegant strategy to novel metal complexes where the ‘classical’ [Ru(bpy)2]2+ core is coordinated to unique large surface ligands [30]. The approach takes advantage of metal coordination to facilitate a dehydrogenation reaction, where extended unfused bpy-type ligands (e.g., 16) are forced into a syn orientation within octahedral complexes (e.g., 17 in Fig. 7). Dehydrogenation at elevated temperatures fuses the fluxional heterocycle into an extended planar ligand yielding a complex with a coordinated large-surface chelate (e.g., 18). It is plausible that the enforced proximity and enhanced steric repulsion within the bound ligand facilitate the single carbon–carbon bond-forming step during the dehydrogenation/cyclization reaction. Since the bpy-type ligands (e.g., 16) can be prepared from two identical or different heterocycles (e.g., 15), the synthesis of symmetrical and asymmetrical coordinated ligands can be achieved (Fig. 7). This modular strategy represents a new facet of our ‘coordination compounds as building blocks’ approaches and can be used for the preparation of unique complexes of hitherto unknown chelating heterocyclic ligands [30].

Dehydrogenation reactions yield novel coordination compounds containing extended ‘large-surface’ polypyridine ligands. The approach takes advantage of metal coordination to facilitate the dehydrogenation/cyclization reaction, where an extended unfused bpy-type ligand (16) is forced into a syn orientation within an octahedral complex (17).
5 Metal-containing biopolymers
Our group has been exploring new approaches for the fabrication of hybrid molecules composed of biopolymers and inorganic complexes. We have utilized the nucleophilic substitution and cross-coupling reactions discussed above for the synthesis of metal-containing amino acids and nucleosides. These useful building blocks have been utilized for the preparation of modified peptides and oligonucleotides, respectively. These, in turn, have been employed for exploring energy and electron transfer processes in metal-modified biopolymers. This section briefly outlines the synthetic aspects of this chemistry and highlights some of the photophysical studies.
Nucleophilic substitutions were used for the single-step preparation of metal-containing amino acids and peptides. Direct substitution reactions between 6 and the thiolate or phenolate side chains of cysteine and tyrosine, respectively, yielded the corresponding modified amino acids (e.g., 19 in Fig. 8) [31]. Since these substitution reactions occur with complete retention of configuration at the metal center [29], diastereomerically-pure metal-containing amino acids and peptides are obtained in one step (e.g., 19, 20) [31]. It is worth noting that such substitution reactions can be performed with protected as well as unprotected amino acids and peptides. Suitably protected derivatives can be further condensed with other appropriate peptidic building blocks to afford short metal-containing peptides [31].
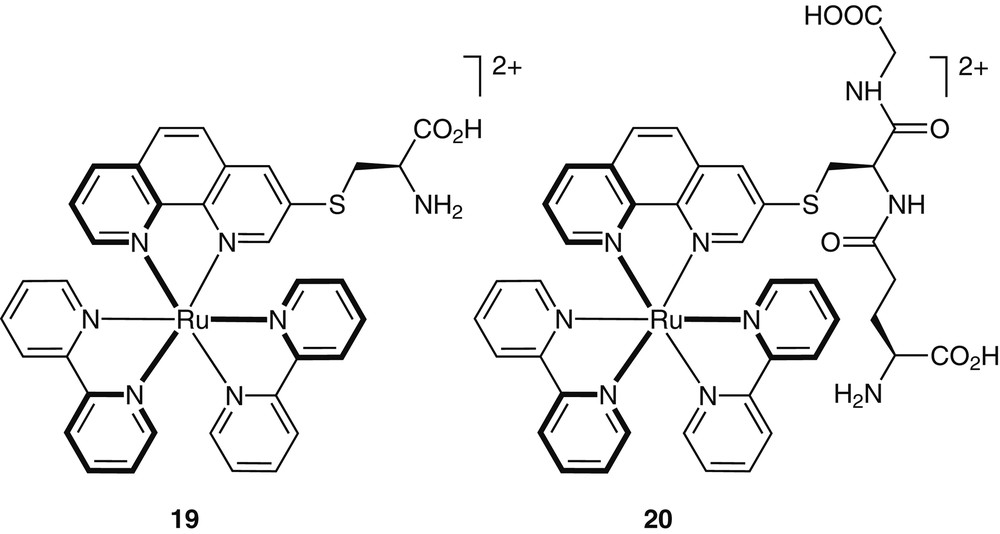
Examples for metal-containing amino acids and peptides can be made in a single step using aromatic nucleophilic substitution reactions with functionalized complexes.
The cross-coupling reactions described above provide a convenient route to numerous metal-containing nucleosides (21–23, Fig. 9). Typically, alkyne-containing nucleosides, such as 5-ethynyl-2′-deoxyuridine, are efficiently coupled in the presence of Pd(0) to halogenated coordination complexes [32]. Interestingly, the alkyne moiety linking the metal complex to the nucleoside’s heterocycle can be hydrogenated to the saturated two-carbon chain (as in 24). These modified nucleobases can be converted into their corresponding phosphoramidites, monomers suitable for automated DNA synthesizers. The availability of such building blocks allowed us to develop a novel methodology for the sequence-specific incorporation of photo- and redox-active metal centers into DNA oligonucleotides using standard solid-phase DNA synthesis [32,33].
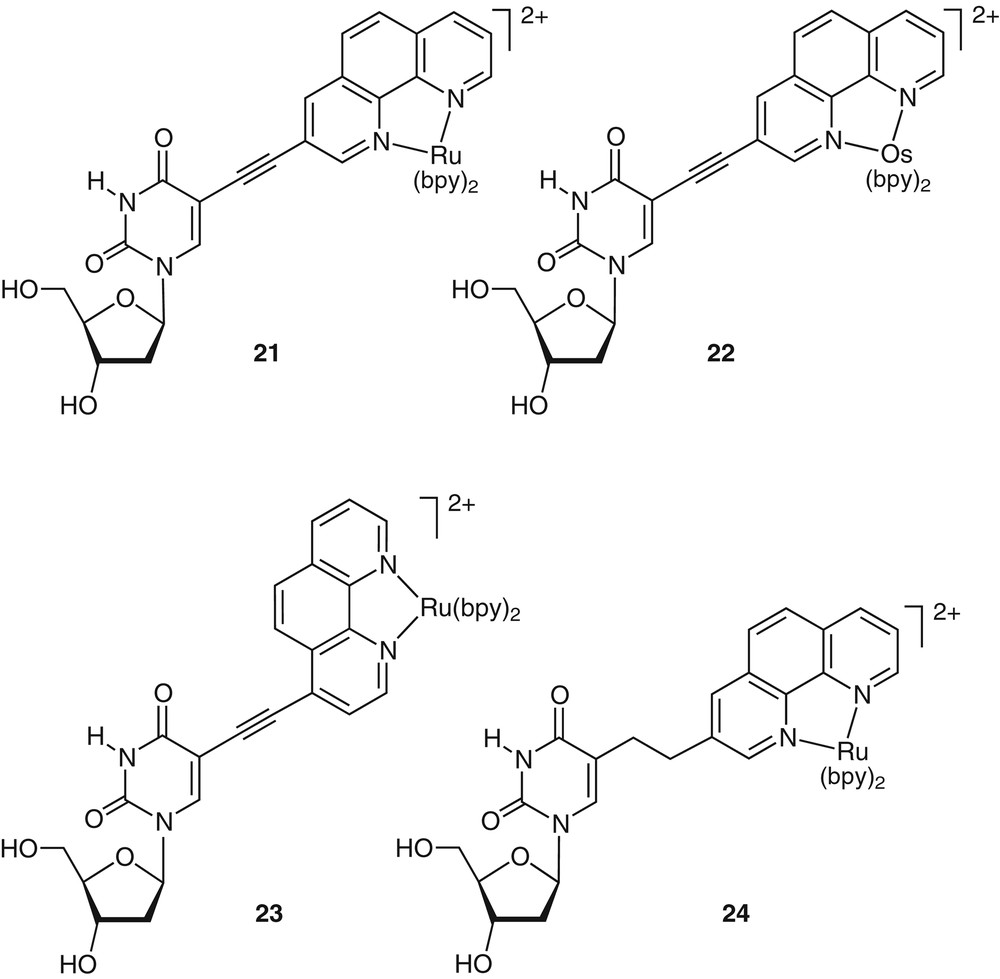
Representative examples of metal-containing nucleosides. Note the versatility in metal ions, linking positions and electronic conjugation.
Metal-containing oligonucleotides, in particular RuII/OsII DNA-bridged diads, are useful tools for the study of photophysical processes in DNA [34]. To systematically investigate energy and electron transfer processes on DNA, we design modified DNA duplexes where the internal positioning of the metal probes within a single DNA template is varied. A series of six doubly modified duplexes that contain the RuII donor 21 and OsII acceptor 22 is shown in Fig. 10. This set of doubly modified oligonucleotides was compared to an analogous series where nucleoside 21 is replaced with 24, its ‘saturated’ analog. Steady-state and time-resolved luminescence spectroscopies of these series of metal-containing duplexes have been utilized to investigate the quenching of the excited donor by the distal acceptor [34]. Detailed analyses of the data have led to the conclusion that the double helix serves as an electronically indifferent scaffold for mediating dipole-dipole Förster energy transfer (particularly for D/A separation between 20 and 50 Å). Deviations from idealized behavior were attributed to DNA dynamics and end effects that may facilitate closer contacts between the D/A pair, thus leading to the possible involvement of Dexter electron exchange processes at short D/A separations [34]. We have also shown that similar metal-containing hairpins sequences can be used as hybridization probes or ‘gene-sensors’ [35]. These observations suggest new opportunities for the utilization of metal-decorated DNA oligonucleotides as novel tools for molecular biological applications.
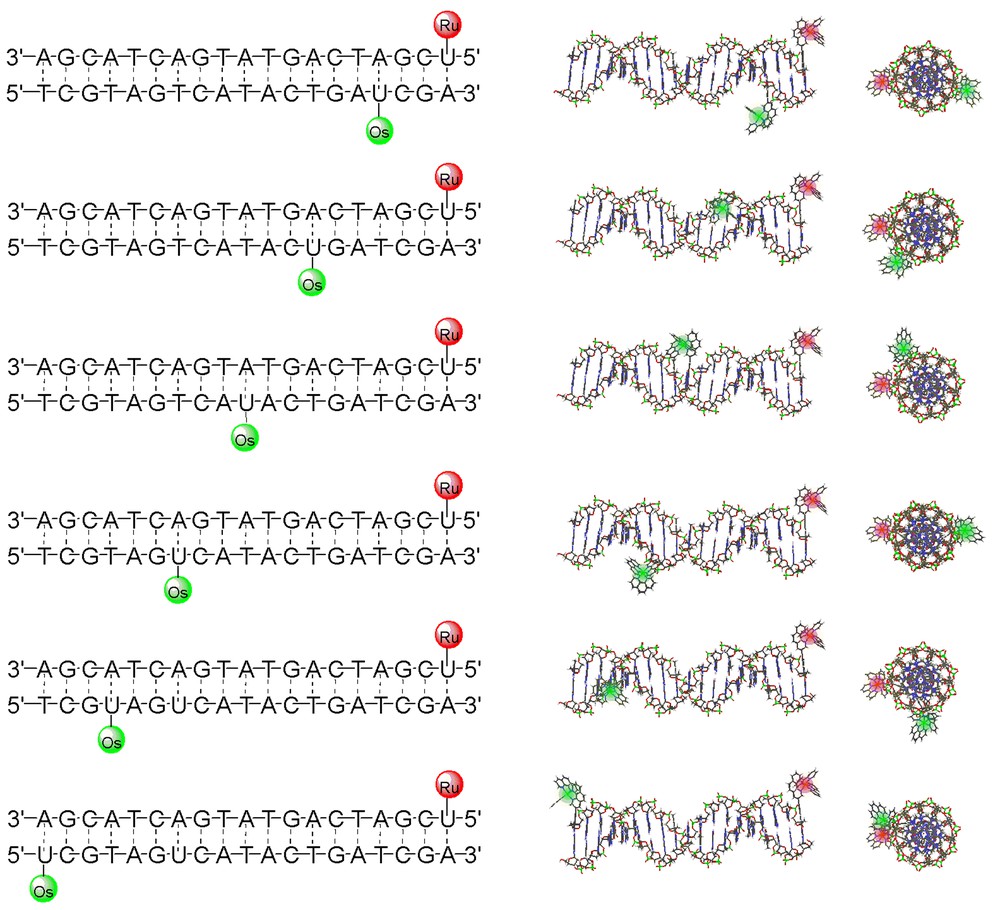
Systematically metal-modified oligonucleotides that have been prepared for investigating the effect of distance on donor-acceptor interactions. In this particular case, the red- and green-labeled metal-containing nucleosides are 21 and 22, respectively. Also shown are side- and top-view models of the doubly modified oligonucleotides.
Our laboratory has also explored enzymatic methods for the incorporation of coordination compounds into nucleic acids [36]. This particular direction has been inspired by the interest in non-optical DNA detection techniques that are amenable to miniaturization (mostly for DNA diagnostic tools). Kinetically inert polypyridine Ru2+ and Os2+ complexes, similar to the complexes described above, are attractive DNA tagging agents due to their chemical stability and favorable redox properties: the Mn/n+1 couples are metal-centered, reversible and their E1/2 can be tuned by the selection of the coordinated ligands. The oxidation potential of [Ru(bpy)3]2+ and [Ru(phen)3]2+ (E1/2 ~ 1.25 V/SCE) is, however, incompatible with ‘inert’ DNA tagging, if nucleotide redox chemistry is to be avoided. If one of the polypyridine ligands, however, is replaced by a negatively charged ligand (e.g., an acetylacetonate or hydroxamate), the resulting complexes [(bpy)2Ru(L)]+ exhibit appreciably lower oxidation potentials, well within an adequate range [37]. We have recently completed the design and synthesis of multiple RuII- and OsII-containing tags with diverse and ‘tunable’ redox potentials that maintain similar structural and chemical features [36]. Importantly, we have synthesized redox-active nucleoside triphosphates that can be enzymatically incorporated at any position by DNA polymerase (e.g., 25, Fig. 11). These results demonstrate that octahedral RuII and OsII complexes are viable, diverse and tunable redox-active tags for DNA modification. The ability of DNA polymerase to efficiently incorporate the metal-containing nucleotide triphosphate suggests the potential use in DNA sequencing and diagnostics.
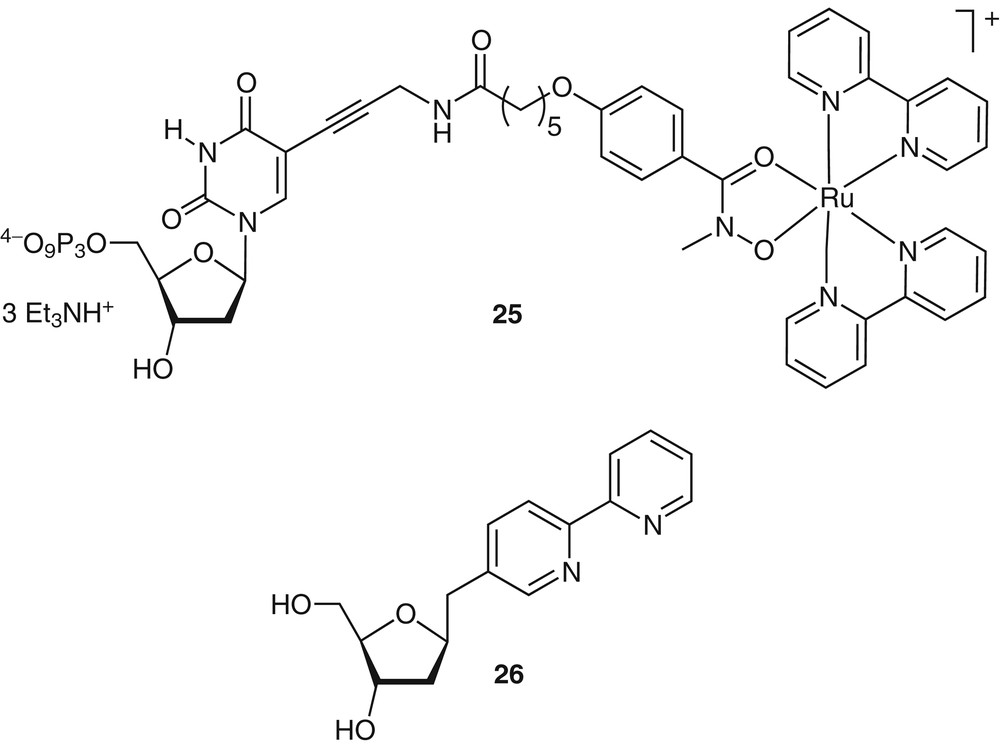
An electrochemically active triphosphate 25 has been shown to be enzymatically incorporated into DNA using the Klenow fragment of DNA polymerase. Also shown is the structure of a bpy ligandoside 26, capable of forming metal complexes within the interior of the double helix.
An alternative approach was taken for the incorporation of metal ions into the interior of the DNA double helix. For this purpose, nucleoside mimics, termed ligandosides, where a metal binding site replaces the heterocyclic base, were designed and synthesized (see 26 in Fig. 11). Incorporation of ligandosides on opposite strands of a DNA duplex can form an inter-strand metal complex. This results in metal-directed pairing rather than hydrogen bonded base pair. We have selected a bpy derivative, a challenging aliphatic C-nucleoside, as our first generation ligandoside [38]. Solid-phase DNA synthesis provided ligandoside-containing oligonucleotides. We were able to demonstrate the formation of an intraduplex Cu2+ complex thus establishing the incorporation of a metal complex into the interior of the double helix [38]. We anticipate such buried metal complexes to assist in deciphering the role played by the π-stacked base pairs in mediating energy and electron transfer processes through DNA. Ligandosides have also been shown to be useful building blocks for the formation of helicates [39].
6 1,10-phenanthroline-based hyperbranched assemblies
The availability of strategically activated phenanthroline derivatives (e.g., 5) has also facilitated other intriguing projects in our laboratory. We have developed an attractive route for the nucleation of hyperbranched assemblies using metal ion coordination (Fig. 12). Novel 1,10-phenanthroline ligands, symmetrically substituted at the 3,8-positions with branched multifunctional groups, were shown to assemble into dendritic-like structures upon metal complexation [40]. In this way, the polyfunctional core is replaced by a coordinated metal ion and the pre-made dendrons are assembled to generate a globular structure [41]. The symmetry and the fractal characteristics of the resulting structures are governed by the coordination number and geometry of the selected metal ion, as well as by the degree of branching of the disubstituted phenanthroline ligand. Depending on the metal ion selected, the resulting dendritic structure may be kinetically labile or kinetically inert.
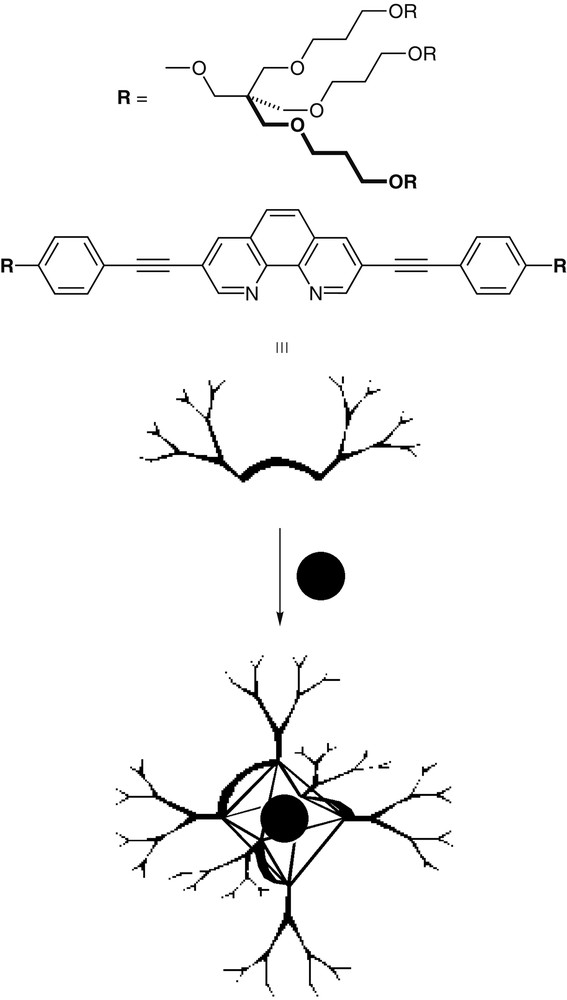
A 1,10-phenanthroline ligand, symmetrically substituted at the 3 and 8 positions with branched multifunctional groups, is assembled into an octahedral dendritic-like structure upon metal complexation. Note that other geometries are accessible by selecting different metal ions.
7 1,10-phenanthroline-based tunable fluorophores
Highly fluorescent 1,10-phenanthroline derivatives have been obtained by extending the conjugation of the parent ligand along the 3–8 axis (Fig. 13). The combination of a coordinating diimine functionality and an extended conjugation via ethynyl linkages provides a unique family of tunable intrinsic fluorophores 27, where changes in the electronic polarization, solvation and coordination are translated into dramatic spectral changes [42]. The emission wavelength of any derivative is dictated by the nature of its substituent and can be further modulated by solvent polarity and by exogenous additives such as protons or metal ions [42]. These observations have prompted us to explore ligand-containing nucleosides as potential ‘reporter’ probes for the local environment of the DNA double helix (e.g., 28, Fig. 13) [43]. The resulting fluorescent DNA oligonucleotides show changes in emission upon hybridization, and are sensitive to the formation of a Watson-Crick vs. mismatched base pairing [43].
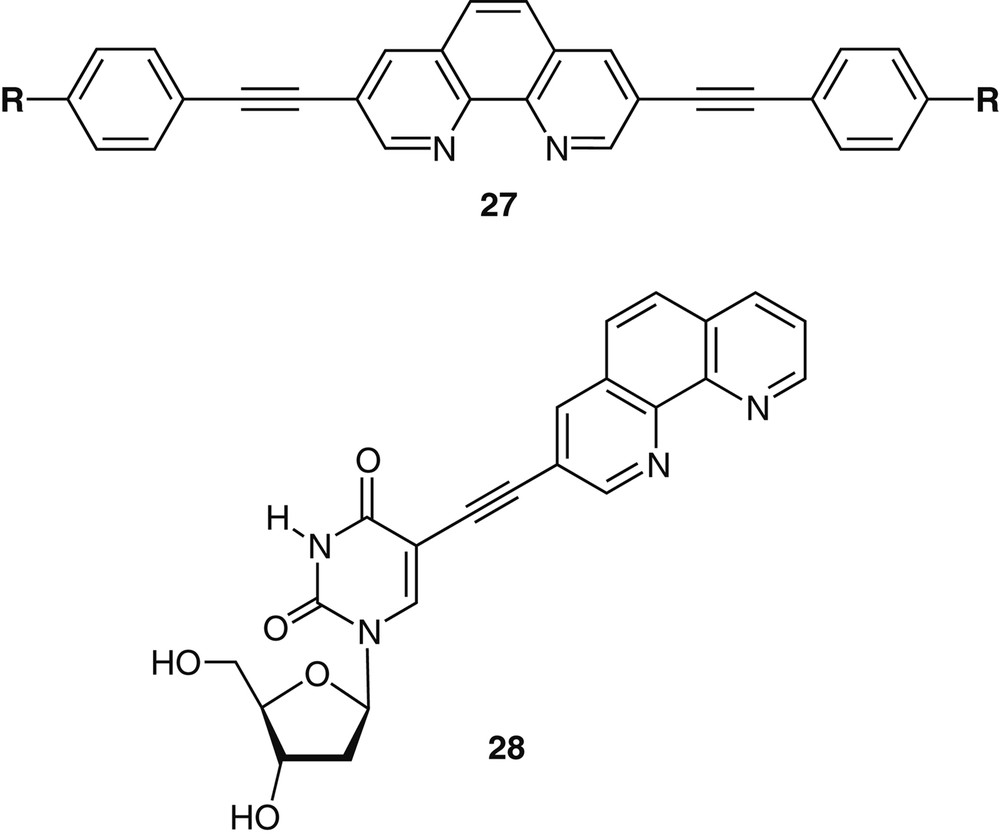
Conjugated phenanthrolines 27 as tunable fluorophres (top) and a fluorescent nucleoside 28 as a hybridization probe.
8 Summary
The incorporation of metal ions into materials is not new [1]. Nowadays, however, we recognize the tremendous structural and functional diversity offered by coordination compounds and their impact on the physical properties upon incorporation into materials. In these early years of the 21st century, coordination compounds enjoy a marvelous renaissance. It appears that chemists have rediscovered metal complexes and exciting new directions and applications are rapidly emerging. As functional molecules, assemblies and materials are taking their place at the scientific frontier, it is only natural to expect scientists to be inspired by such a flourishing field as coordination chemistry. Indeed, coordination compounds are now found in almost every branch of modern chemistry. It is likely we will continue and see innovative utilizations of coordination compounds in materials research for years to come.
Acknowledgements
I thank my co-workers for their contributions (see references) and for facilitating the evolution of this chemistry. I am grateful to Motorola Clinical Micro Sensors, the UC BioSTAR Project and the National Institutes of Health (Grant Number GM 58447) for their generous support.