

1 Introduction
Research progress in dendrimer chemistry is picking up in a tremendous pace due to the unique structural features of these aesthetically pleasing macromolecules [1]. The highly branching nature of dendrimers creates a special class of molecules that possess unusual properties that are not often observed in random coil polymeric systems. The presence of a large number of surface groups in a dendritic skeleton can generate several special effects. First, the close proximity of the large number of surface groups may induce positive or negative allosteric interactions. Secondly, the physical shielding/blocking effect of surface groups may also produce a change of the physical/chemical properties of the internal functionalities. Thirdly, recent advances in dendrimer synthesis now enable us to prepare dendritic macromolecules having segregated internal microenvironment bearing different physical properties; this could also produce supramolecular dendritic systems with interesting properties. All of these effects, also known as the ‘dendritic effects’, are unique dendritic systems and are the subject of interest in this article.
2 Dendritic effects due to functional-group multiplicity
Perhaps the first dendritic effect ever disclosed was originated from a reactivity study of multi-center catalytically active dendrimers (e.g., 1) as reported by Ford [2]. It was found that the catalytic reactivity of the decarboxylation reaction of 6-nitrobenzisoxazole-3-carboxylate was not directly proportional to the number of catalytic centers on the dendrimer surface. The G2 polyammonium dendrimer was 10 times catalytically more reactive than the corresponding G1 analog on a per catalytic site basis. This was attributed to a much higher local density of the quaternary ammonium ions in the G2 dendritic surface that created a much more hydrophilic microenvironment for the rapid decarboxylation. A similar positive catalytic dendritic effect was also observed by Detty in his study of dendritic polyphenylselenides [3]. Positive catalytic dendritic effects were also noted by Jacobsen [4], in which the higher generation Co(salen) catalysts (e.g., 2) were far more effective than the lower ones due to interchain cooperative interactions among the catalytic sites located on the dendrimer surface. Ironically, such interactions among catalytic sites are not always beneficial. For example, van Koten reported a case wherein the higher generation carbosilane dendritic catalysts became inactive due to deactivation arising from multi-site interactions [5] (Fig. 1).
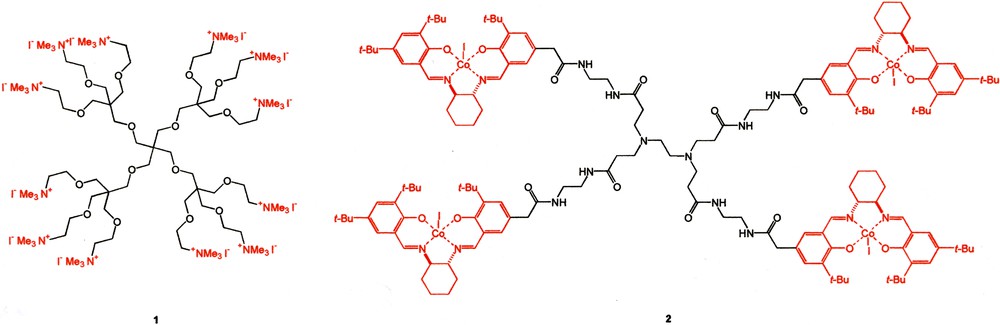
Surface-group multiplicity can also be used to construct dendritic receptors with enhanced binding affinity for guest molecules. For example, Astruc reported a notable dendritic effect on the binding of Cl– anions to polyamidoferricinium dendrimers (e.g., 3) [6]. The G2 dendrimer had a much higher affinity (10 ×) towards Cl– than the analogous G1 compound, which in turn exhibited ten times higher affinity than the G0 analog. NMR titration experiment showed that the binding involved the interaction between Cl– and a tripodal dendrimer branch. It was reasoned that the higher sterically congested environment in the G2 compound forced the tripod branches to adopt a better-sized cavity for enhanced Cl– recognition. Our group also reported the enhanced binding of protic solvents by high-generation β-alanine-based dendrimers 4 [7]. This was due to the presence of a greater number of amide and carbamate functional groups in the G3 dendrimer that greatly enhanced the binding strength towards protic solvents. This then led to an observed enhancement of H/D exchange rate of the amide N–H in the higher generation compounds (Fig. 2).

Vögtle also reported the preparation of a G4 poly(propyleneimine) dendrimer 5 having 32 fluorescent dansyl sensor units at the surface and 30 aliphatic amino groups in its interior [8]. This compound functioned as a highly efficient Co(II) ion sensor with significant signal amplification due to fluorescent quenching of all the dansyl units even when only one of the amino groups was complexed to Co(II) (Fig. 3).

Another useful design making use of the surface multiplicity nature of dendrimers is the synthesis of highly efficient dendritic light harvesting systems. A large number of photochemically reactive groups on the surface periphery can act as antennas to absorb light energy and transfer it to the central core. One such system was reported by Moore, in which the photochemically reactive functionalities were constructed on the periphery of a poly(phenyleneethynylene) dendritic skeleton while the central core had a perylene focal point group (e.g., 6) [9]. Excitation of the phenyleneethynylene dendrons resulted in energy transduction to perylene. The light harvesting ability of these compounds increased with increasing generation due to an increase of the number of energy collecting sites. However, the quantum yield of the energy transfer decreased with increasing dendrimer generation. Laser–dye-labeled poly(aryl ether) dendritic antennas having similar light harvesting efficiency profile had also been reported by Fréchet [10] (Fig. 4).

Recently Aida reported that the quantum yield of the energy transduction process was strongly dependent on the dendrimer morphology [11]. Among a series of porphyrin dendrimers substituted with different number of G5 polyether dendrons, the tetrasubstituted dendrimer 7 having a structurally rigid, spherical geometry showed a much higher energy transfer quantum yield than partially substituted compounds (e.g., 8). This was attributed to the fact that the four closely-packed dendron subunits in compound 7 created a continuous array of chromophores to allow the excitation energy migrate efficiently over the dendrimer skeleton and not being trapped in localized regions as compared to the partially substituted dendritic porphyrins.
3 Dendritic effects due to steric shielding or blocking
The physical size of the dendritic shell has the ability to act as a shielding shelter and may therefore confer extra stability for labile functionalities sitting inside the interior. For example, Aida showed that the half-life of thermally unstable bis(μ-oxo)dicopper complexes 9 could be prolonged by appending polyether dendritic fragments to the dicopper core [12]. Hence, the half-lives of the G1–G3 complexes were 7, 24 and 3075 s at –10 °C, respectively. Thermodynamic studies indicated that there was a much larger entropy loss required for the intramolecular oxidative self-decomposition for the G3 complex as compared to the lower generation analogs. This was due to the difficulty in reorganizing the reactive partners in the sterically highly congested G3 complex. Similarly, a Fe(II)–porphyrin 1-methylimidazole complex covalently encapsulated within a G5 polyether dendritic cage displayed reversible oxygen-binding activity whereas the corresponding complex within a G1 dendritic shell rapidly and irreversibly oxidized in an oxygen atmosphere [13] (Fig. 5).

The physical size of the dendritic surface may also act as a shield to retard certain physical/chemical processes of the internal functionalities. For example, it was demonstrated by Newkome [14] and us [15] that the redox processes of an electrochemically active dendrimer core (e.g., 10) became increasingly irreversible due to the hindrance of electron transfer between the electrode surface and the buried redox-active unit by the increasing size of the dendrons. Furthermore, redox potentials can also be shifted depending on the nature of the appending dendron and the solvent medium. Smith recently reported an in-depth study of the dendritic effects on the redox potential of core-centered ferrocene dendrimers (e.g., 11) and showed that in ‘good’ solvents where the dendrons could adopt an open structure, the dendritic branches appeared to have little effect on the redox potential [16]. On the other hand, in ‘poor’ solvents where the dendrons had a relativity compact structure due to insufficient solvation, the core was more effectively shielded. Attachment of dendritic branches to the electrochemically active ferrocene unit therefore resulted in an increase of oxidation potential due to the destabilization of the oxidized ferricenium state by the less polar dendrons as compared to the more polar surrounding electrolyte solution. In general, dendritic branches operated by shielding the core from the relatively polar, ionic electrolyte solution, preventing ion-ion interactions, and thus hindering the generation of a charged species. However, an exception to this trend had been observed by Kaifer, in which dendritic branching actually facilitated the oxidation process in a series of ferrocene-cored unsymmetrical dendrimers (e.g., 12) [17]. It was believed that in this case the redox active center was capable of orienting itself at the electrode surfaces to allow facile electron transfer due to the asymmetric nature of the dendrimer. Hence, in this particular case the ferrocene group was not completely isolated from the surrounding medium (Fig. 6).

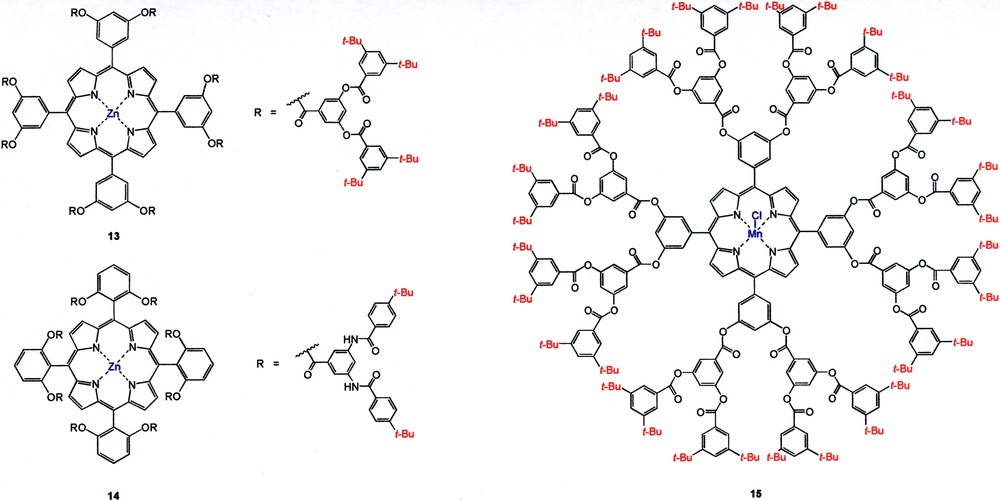
Another interesting application of the dendritic steric shielding effect was the design of shape-selective host molecules as reported by Suslick [18]. A series of dendritic Zn-porphyrin receptors having different substitution patterns was prepared in which the Zn binding site was shielded by aromatic polyamide/polyester dendrons. Their binding strengths to various nitrogen-based ligands of different size and shape were then investigated. First, all pyridine-based ligands, irrespective of size and shape, had a much higher binding constant (10 ×) to the higher generation 3′,5′–substituted Zn-dendritic porphyrins (e.g., 13) than to the one without dendritic appendages. This was due to favorable π-π interactions between the pyridine moiety and the aromatic dendrons. Second, the 2′,6′–substituted dendritic porphyrin 14 was highly selective hosts for linear amino ligands (Keq ~ 104 M–1), while branched amino compounds such as nicotine and quinine were poor ligands (Keq ~ 100–10–2 M–1). Based on modeling study, the 2′,6′–substituted dendritic porphyrin had a much narrower cleft for binding to the branching amino ligands. Hence, dendritic molecules can act as substrate selective receptors such as those exist in biological systems.
An interesting catalyst design making use of the shielding effect was reported by Moore in the substrate selective epoxidation of olefins catalyzed by core-centered Mn-based porphyrin dendrimers (e.g., 15) [19]. The higher generation G2 catalyst, having a sterically more congested active site, preferentially reacted with sterically less hindered olefins. On the other hand, a non-dendritic Mn-porphyrin analog showed little substrate selectivity (Fig. 7).
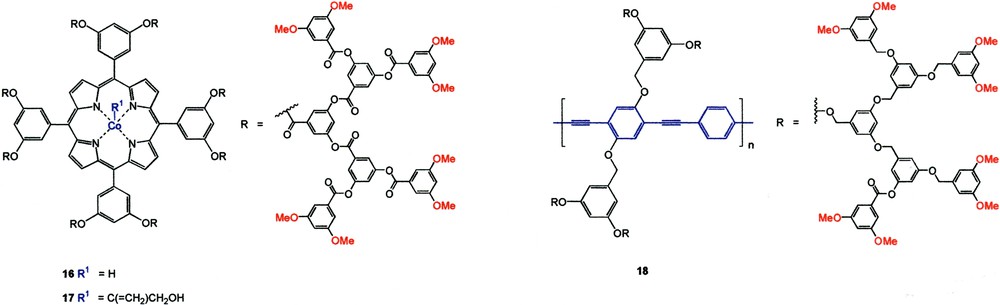
The dendrimer cage can also be used to protect reactive chemical species from undergoing further transformations [20]. (Porphinato)Co–H dendrimers (e.g., 16) were known to react with propargyl alcohol to form the corresponding organocobalt(III) species (Co[III]–C[=CH2]CH2OH 17). In the absence of dendritic shell protection, compound 17 underwent intermolecular-induced isomerization to Co(III)–C(CH3)=CHOH or Co(III)–CH(CH3)CHO. However, when the (porphinato)Co–H core was encapsulated inside a tetrasubstituted G3 polyester dendron, isomerization failed to occur due to steric protection by the cage that prevented the access of another cobalt porphyrin molecule (Fig. 8).
Dendritic shielding effect may also be used to prevent collisional quenching of photoexcited states of luminescent compounds as reported by Aida [21]. Poly(phenyleneethynylene) rod polymers 18 decorated with G4 polyether dendrons showed strong blue fluorescence at 454 nm with a quantum yield of nearly 100%. Reducing the size of the decorative coat to G3 or G2 led to a gradual decrease of quantum yield efficiencies. In addition to preventing intermolecular collisional quenching, the polyether dendrons also improved the solubility of the rigid poly(phenyleneethynylene) backbone (Fig. 9).
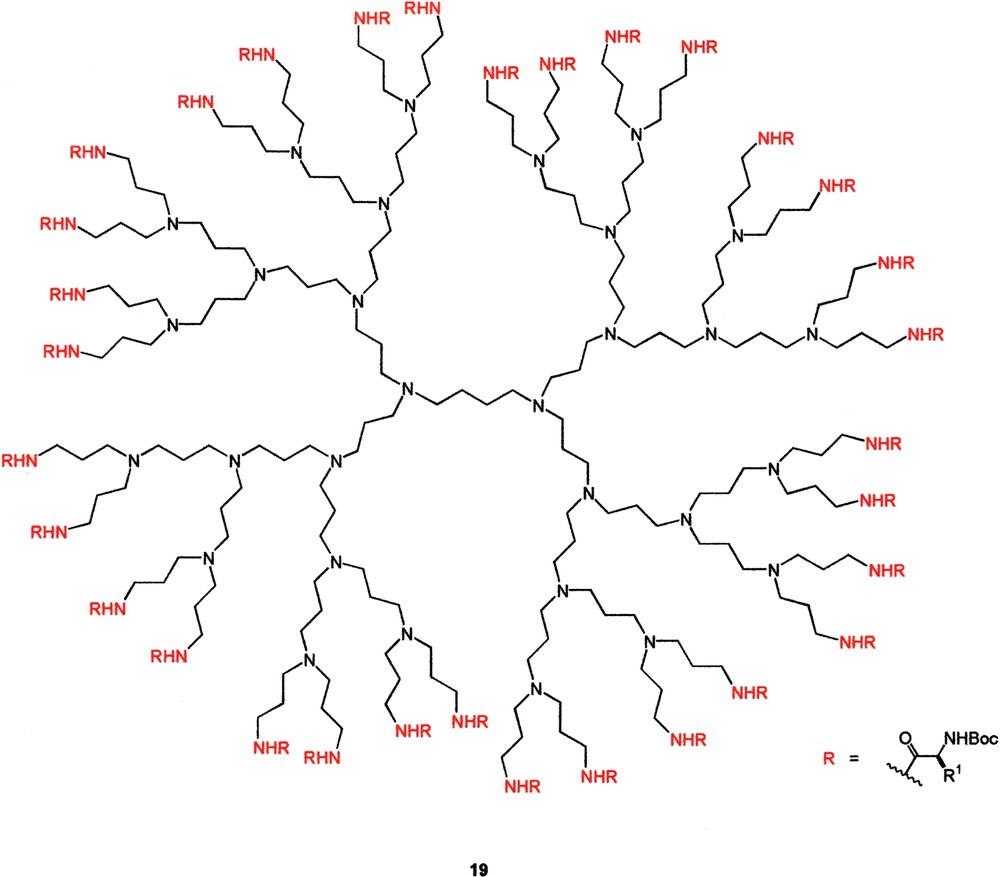
One intriguing ‘dendritic effect’ was revealed by Meijer in his chiroptical study of poly-(propyleneimine) dendrimers (e.g., 19) having the surface decorated with Boc-protected amino acid residues [22]. It was found that both the specific rotation and Cotton effect diminished from the G1 to G5 dendrimers when the side chain of the amino acid moiety was relatively bulky. This finding was unexpected as most chiral dendrimers reported in the literature showed a linear relationship between the chiroptical properties and the number of chiral units resided within the dendritic skeleton [23]. Careful examination revealed that this series of dendrimers, especially the higher generation ones, had a very rigid shell because of the presence of a large number of interchain hydrogen bonds arising from the close proximity of the amide and carbamate groups. As a result, the chiral amino acid residues on the dendrimer surface had a number of frozen-in conformations, which yielded an average chiroptical property of almost zero. This effect was more prominent for end groups whose chiroptical property was highly sensitive to the local environment (Fig. 10).

The interesting consequence of having a rigid shell structure together with the presence of internal voids inside a dendrimer resulted in the formation of dendritic boxes that could encapsulate guest molecules. For example, when a flexible G5 amino-terminated poly(propyleneimine) dendrimer 20 was end-capped with N-Boc-phenylalanine groups to produce a rigid shell dendrimer 21 in the presence of 3-carboxy-proxyl or eriochrome black T, physical encapsulation of these guest molecules occurred [24]. Interestingly, dendrimers of lower generations such as G3 were not capable of trapping guest molecules; their surface domains were not dense enough to form a closed shell. Meijer also showed that guest molecules of different sizes and shapes could be selectivity liberated after being encapsulated into the N–Boc–phenylalanine-capped poly(propyleneimine) dendrimer 21 [25]. After trapping a mixture of rose bengal and p–nitrobenzoic acid molecules into the dendritic structure, the Boc groups were then removed by acid hydrolysis to produce a perforated dendritic box having a less rigid shell. It was found that the smaller p–nitrobenzoic acid molecules could diffuse out into the solution while the larger rose bengal molecules were still retained inside the dendritic framework. This ability to fine-tune the permeability of a dendritic shell offers an exciting way to produce selective drug delivery systems.
4 Dendritic effects due to changes in the nature of internal microenvironment
Instead of modifying the size of the surface blocking groups to trigger the release of encapsulated molecules, one can also change the internal microenvironment of a dendrimer to produce controlled release systems [26]. For example, pyrene was shown to bind strongly to G4 or G5 amino-terminated poly(propyleneimine) dendrimers (e.g., 20) in aqueous solutions at pH ~ 11. Upon addition of hydrochloric acid, the amino groups inside the dendrimer were protonated. The internal environment became highly polar, which tended to repel the hydrophobic pyrene molecules and forced their release into the solution. In this example, the guest molecules were not encapsulated but were bound to the internal functionalities via non-covalent interactions. Another pH dependent guest delivery system involving hydroxy-terminated PAMAM dendrimers was also reported by Twyman [27].
Vinogradov also reported that a change of the solvent medium could alter the ‘porosity’ of the dendrimer shell and the O2 dependent phosphorescence quenching behavior of a Pd-porphyrin core [28]. Pd-porphyrin dendrimers (e.g., 22) decorated with carboxylated terminated poly-L-glutamate dendrons were prepared. In DMF as the solvent, where the dendritic arms were fully extended due to effective solvation, there was almost no difference in the oxygen quenching constants on the phosphorescence of the porphyrin core for dendrimers of different generations. On the other hand, in water solution the dendritic arms shrank and formed more densely packed cages around the central core. The diffusion of O2 into the phosphor core was therefore impeded by the larger dendrons and hence the quenching behavior in aqueous solution was strongly dependent on the dendrimer generation (Fig. 11).

Smith recently disclosed the synthesis of dendritically modified tryptophan derivatives (e.g., 23) and showed that the polyamide–polyester dendrons had a profound effect on the fluorescence property [29]. For the NH (23, R = H) series in non-hydrogen bonding solvents, the NH group of the indole ring was hydrogen bonded to the G2 polyamide–polyester dendrons due to back folding. This led to a red shift in emission wavelength of the tryptophan core for the G2 dendrimer as compared to the G0 analog. However, in hydrogen bond donor or acceptor solvents, the NH functionality was solvated to the same extent in all the G0–G2 dendrimers, and hence there was only very little red shift in emission upon dendrimerization. On the other hand, for the N–Me (23 R = Me) series, interactions of N–Me with either the dendritic branches or the solvent were not possible due to its inability to form hydrogen bonds, and hence no shift in emission frequency could be observed in hydrogen bond solvents.
By introducing polar alcoholic functionalities in the interior of hydrophobically shelved dendrimers (e.g., 24, X = CH2OH), Fréchet was able to enhance their catalytic activity towards the unimolecular E1 elimination of a tertiary iodide [30]. The long hydrophobic hydrocarbon chain located on the surface served to simulate the non-polar reaction medium while the interior polar CH2OH groups aimed to stabilize the carbocationic intermediate developed during the E1 elimination. Decreasing the size of the dendrimer from G4 to G3 resulted in a 15–20% reduction in both reaction rate and turnover number. Interestingly, replacing the hydroxymethyl groups by the less polar ester groups (24 X = CO2Me) also resulted in a 10–15% reduction of reactivity (Fig. 12).
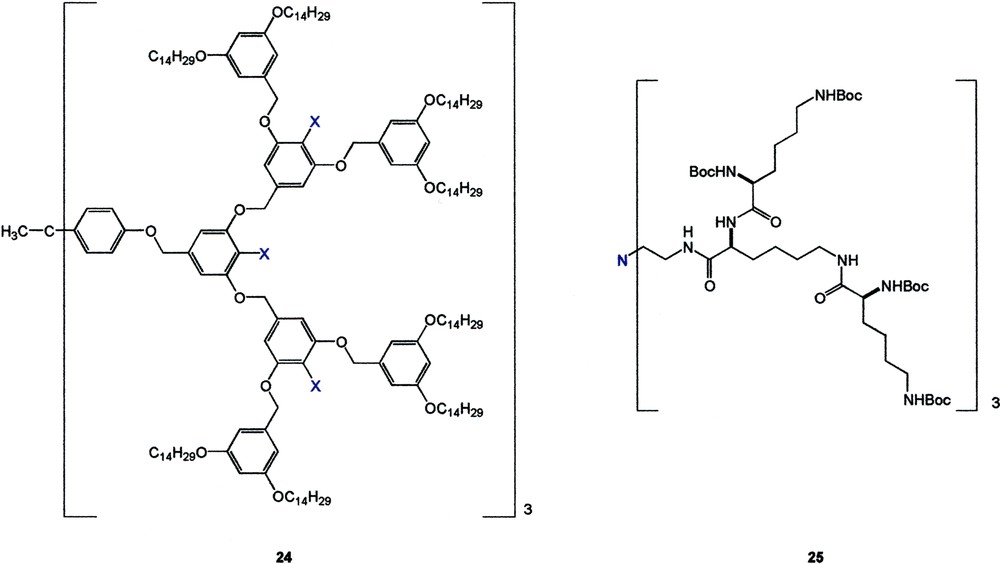
Smith also reported that the G2 poly-l-lysine-based dendrimer 25 having a catalytically active amino core could provide a much more polar, hydrogen-bonding environment than the corresponding G1 analog for the anionic nitroaldol reaction between 4-nitrobenzaldehyde and nitroethane [31]. Due to steric shielding, core-centered single site catalysts generally show a gradual decrease of reactivity with increasing dendrimer generation [32]. However, the presence of more hydrophilic L–lysine moieties in the G2 catalyst could provide better stabilization of the anionic transition state, therefore the reaction was faster as compared to the use of the G1 catalyst (Fig. 13).
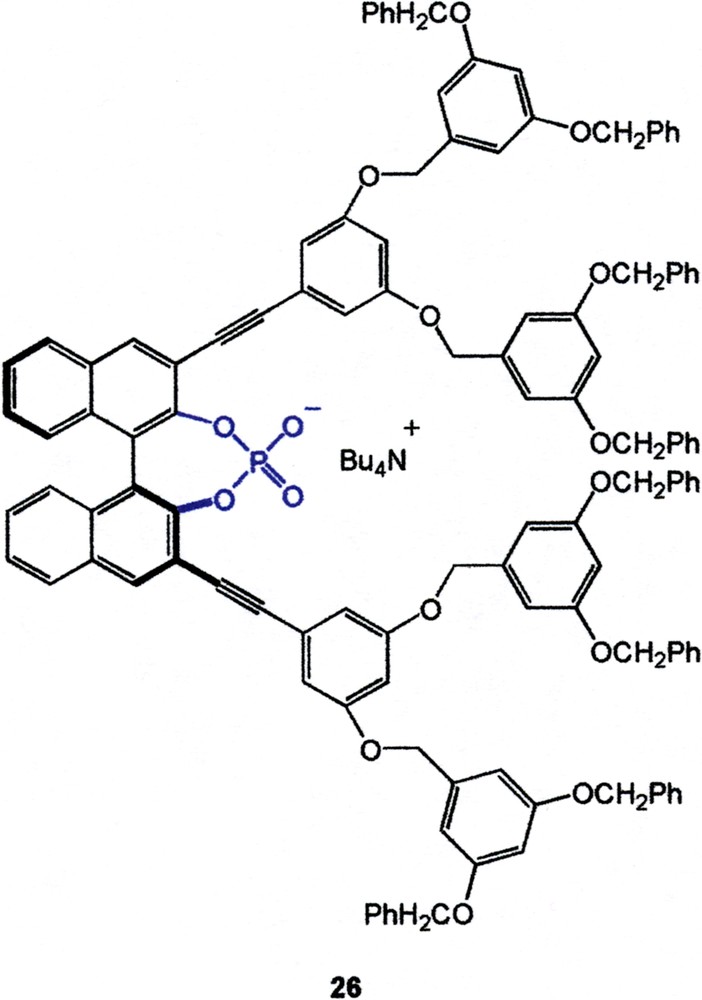
A series of studies by Diederich on dendritically modified hosts having an endo receptor site demonstrated a gradual decrease of binding efficiencies to guest molecules with increasing dendrimer generation [33]. However, there are also examples in which addition of dendrons can lead to a slight increase of binding capability. For example, the binding constants of various octyl glucosides towards dendritic 1,1′–binaphthalene–derived phosphate receptors (e.g., 26) were found to increase gradually from G0 to G2 and then decrease for the G3 analog [34]. Therefore, for the lower generation compounds, a favorable interaction between the hydrophobic dendrons and the hydrophobic octyl side chain was the dominant factor in controlling the binding strength before strong steric repulsion came into effect in the G3 host.
The examples described here are by no means the exhaustive illustration of the various dendritic effects reported in the literature. Nonetheless, they highlight the tremendous potentials of functional dendritic molecules to serve as a new class of molecules for catalysis, material and biological applications. It is certain that novel functional dendritic molecules possessing new and novel dendritic effects will continue to appear in the near future.
Acknowledgements
We thank the Research Grants Council, HKSAR for the financial support (CUHK4273/00P).