

1 Introduction
There has been a continuing need to control the surface chemistry of chemically functional colloids in general, and polymer colloids in particular (the latter, because their size and size distribution are relatively easy to control by means of emulsion polymerization). Such colloids have found important uses in biological cell separation and identification, catalysis, cancer chemotherapy, DNA and RNA probes, and in next-generation computer technologies, to name a few. Of course, by far the largest use of polymer colloids is in the realm of construction materials, e.g. latex paints, adhesives, caulks, and elastomeric cementitious and gypsum-based products. Some of these traditionally have been plagued by poor adhesion and poor water-resistance because of surfactant migration upon drying. If the surfactant is bound to the particle surface, then not only can it not migrate to interfaces, but also one needs less of it, which in turn means less water sensitivity. A couple of recent papers confirm these expectations [1,2].
We developed a quantitative method for the determination of surface sulfonate groups by means of secondary ion mass spectrometry (SIMMS), a technique that samples only the first one or two molecular layers, in contrast to almost every other method more commonly employed [3]. Although the SIMMS work was published years ago, the transurf synthetic work upon which it was based was published only in the form of Fifield’s PhD thesis [4]. This paper now sets forth the synthetic work in a more accessible form. Fitch had suggested as early as 1975 that chain transfer could be employed deliberately to regulate surface chemistry of polymer colloids, e.g. by employing mercaptoacetic acid to introduce carboxyl groups to particle surfaces [5]. Fitch and McCarvill found that coincidental chain transfer to ordinary surfactant, e.g. sodium dodecyl sulfate, SDS, or sodium dodecyl sulfonate, SDSo, used to stabilize the latex could be responsible for up to half of the bound surface functional groups [6]. Since that time a great deal of work has been published in this field [7]. This paper deals with a molecule, which at the time of its synthesis was the first transurf, i.e. a surface-active molecule that had a highly reactive chain transfer group. It was anticipated that this could lead to general procedures for the regulation of the surface chemistry of polymer colloids.
2 Sodium mercapto-decane sulfonate (SMDSo)
2.1 Outline of the synthesis
A modified Schramm synthesis [8] was used, starting with 1,10-dibromodecane according to the following scheme:
- 1.
- 2.
- 3.
- 4.
In Step 1, a 1.5 × molar excess of the dibromodecane reacted with sodium sulfite in aqueous ethanol under reflux for 30 h. The monosubstituted product was isolated by solvent extraction and purified by recrystallization. The product of the reaction of this bromoalkanesulfonate with thiourea in aqueous solution under reflux for one hour in Step 2 was the thiouronium bromide salt. It separated as colorless crystals upon cooling, and was recrystallized from ethanol/water (50/50) in 79% yield. This was hydrolyzed in Step 3 to the mercaptan on one end of the molecule, while on the other, guanidinium ion replaced sodium. The sodium was reintroduced in Step 4 by means of ion exchange with H+, followed by titration with NaOH. Because mercaptans can be readily oxidized to disulfide in air, the last steps were carried out in an oxygen-free nitrogen atmosphere. Details are given in the experimental section.
2.2 Characterization
Two qualitative tests, ethanolic lead acetate and sodium nitroprusside, both confirmed that the product of synthesis was a mercaptan. Thin layer chromatography indicated a minor contaminant, which was confirmed to be the corresponding disulfide, no doubt formed by air oxidation of the SMDSo. Quantitatively, titration with standardized ethanolic iodine showed that the product was more than 98% pure SMDSo.
Structure proof was obtained by proton NMR and infrared spectroscopy. In the NMR, peaks at 1.29, 2.4 and 2.8 ppm shifts corresponded to known methylene protons in mid-molecule, adjacent to –SH and adjacent to –SO3–, respectively. In the IR spectrum, absorbances at 720, 790, 1050 (strong, sharp), 1160 (strongest, broad), 1450 (shoulder at 1400) and 1630 cm–1 were confirmatory for SMDSo.
2.3 Surface properties of SMDSo
2.3.1 Determination of CMC
The ultimate test of importance to us was whether, and to what extent, this molecule is surface-active. It should be a micelle-former, and if so, a plot of solution conductance as a function of concentration would show a change in slope at the CMC. This was indeed the case as shown in Fig. 1 . Measurements were made at 20 °C with a Beckman Mysels-type conductivity cell and an Industrial Instruments bridge. The CMC was determined to be 6.1 × 10–2 mol l–1. Sodium decanesulfonate has been found to have a CMC of 4.1 × 10–2 mol l–1. The higher value for SMDSo is expected because of the hydrophilicity of the –SH group.
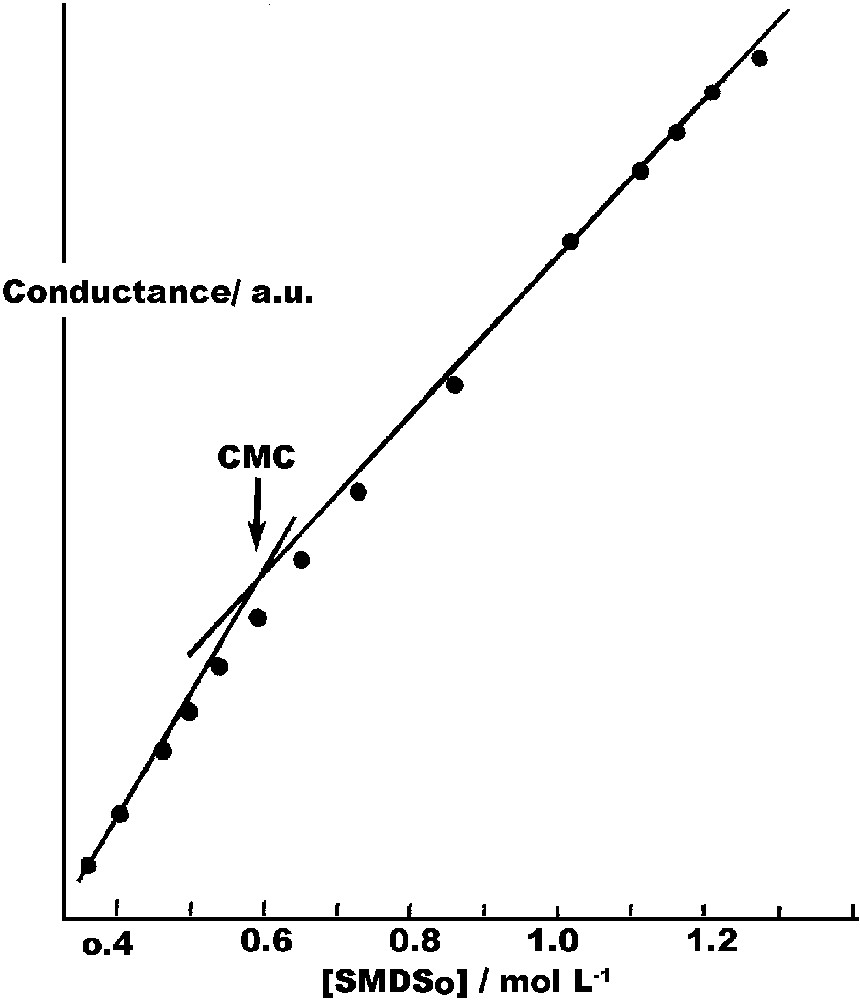
Determination of CMC by conductance.
2.3.2 Surface tension of solutions
Surface tensions, σ, of serial dilutions of SMDSo solutions of concentration C were obtained using Wilhelmy plate technique with a Rosano film balance. Because of the exposure of the surface excess to the atmosphere, we conducted all measurements in an atmosphere of oxygen-free nitrogen. Even so it became clear that some of the highly surface-active disulfide was being formed by oxidation at the interface, so that extreme care was required, and only brief spans of concentration were employed. The minimum in the Gibbs plot shown in Fig. 2 shows the effect of this contamination. Nevertheless the CMC so obtained is the same as that found conductometrically. Fig. 3 is a composite of several experiments in each of which only a few measurements were made to minimize disulfide formation. It is the best estimate of surface tension as a function of concentration, and extrapolates reasonably well to the surface tension of pure water at infinite dilution. Even so, there is some fall-off in σ at very low concentrations, no doubt attributable to disulfide.
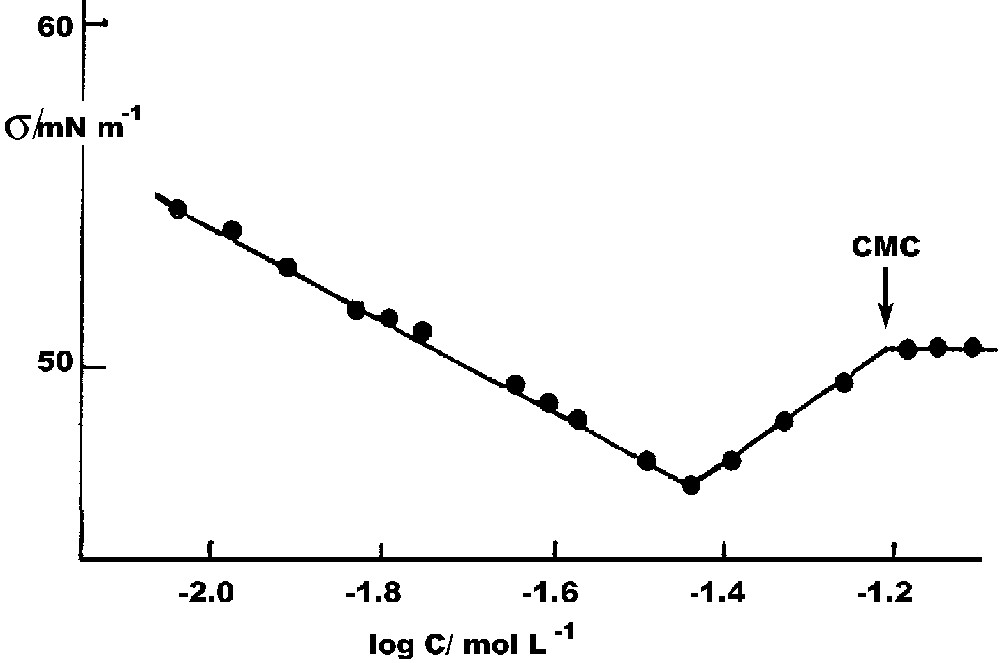
Gibbs plot of SMDSo.

Gibbs plot in dilute region.
2.4 Test for chain transfer during polymerization
A ‘quick and dirty’ experiment was carried out to compare two polystyrene latexes, one of which used sodium dodecyl sulfonate, SDSo, and the other used an equimolar amount of SMDSo. In all other respects the two recipes were the same. The initiator for the emulsion polymerization for both was 4,4′-azobis-(cyanovaleric acid) – AIBN –, which should not react significantly with the mercaptan. After polymerization, both colloids were gently shaken with highly purified, mixed bed ion-exchange resins (in the OH– and H+ forms). This was repeated two more times, at the end of which the SDSo latex had coagulated completely, whereas the SMDSo latex showed no evidence of coagulum. It is well known that ion exchange will remove adsorbed surfactant, so that it is clear that in this case, when the SDSo was removed, there was insufficient surface charge to stabilize the colloid. The SMDSo apparently was grafted to the surface by chain transfer during the emulsion polymerization, and thus could not be removed by the resin. So the colloid made with it remained stable.
2.5 Making polystyrene colloids with SMDSo
A few experiments were carried out to determine whether SMDSo would act as expected for a surface-active agent in emulsion polymerization. Two sets of experiments were conducted: Set I would produce colloids containing only sulfonate surface groups; Set II would form bifunctional colloids containing both sulfonate and carboxyl (Tables 1 and 2).
Set I
| Colloid | B1 | B2 | B3 |
| Water (ml) | 85 | 85 | 85 |
| Styrene (ml) | 5 | 5 | 5 |
| Biacetyl (0.023 M solution) (ml) | 10 | 10 | 10 |
| SMDSo Concentration × 103 (M) | 8.0 | 4.0 | 1.2 |
| Results | |||
| % Solids | 3.43 | 3.07 | 1.04 |
| % Conversion | 70.0 | 64.8 | 21.9 |
| meq H+/g polymer* | 6.08 × 10–2 | 4.42 × 10–2 | 1.57 × 10–2 |
* Surface acid concentration determined conductometrically after ion exchange. Only strong acid was detected.
Set II
| Colloid | C1 | C2 | C3 | C4 |
| Water (ml) | 45 | 45 | 45 | 45 |
| Styrene (ml) | 5 | 5 | 5 | 5 |
| SMDSo concentration (M) | 5.1 × 104 | 9.8 × 10–4 | 5.0 × 10–3 | 1.0 × 10–2 |
| Azocyanovaleric acid | 5.0 × 104 | 5.0 × 10–4 | 5.0 × 10–4 | 5.0 × 10–4 |
| Results | ||||
| % Solids | 2.85 | 3.49 | 6.22 | 6.55 |
| % Conversion | 31 | 38 | 68 | 72 |
| meq. H+/g polymer* | 4.23 × 10–3 | 7.07 × 10–3 | 1.66 × 10–2 | 2.18 × 10–2 |
| Particle Dia. (nm) | 420 | 500 | 440 | 420 |
* Surface acid concentration determined conductometrically after ion exchange. Both weak and strong acids were detected.
Both sets were conducted under oxygen-free nitrogen after scrupulous purging of oxygen. The initiator in Set I was biacetyl (butanedione-2,3), which photolyzes readily with visible light to produce nonionic free radicals, and that in Set II was 4,4'-azobis (cyanovaleric acid) which thermolyzes to split out N2 to form two identical free radicals. Both initiators are water-soluble The results for both sets are shown in Tables 1 and 2.
2.6 Control of surface group concentration
One of our goals in this work was to be able to control the concentration of surface functional groups on polystyrene (PS) colloidal particles independently of particle size. Ordinarily in emulsion polymerization one regulates particle size by the amount of surfactant added during the particle formation stage. But surface group concentration is affected by the concentration of initiator and by the extent of grafting of surfactant molecules onto the particles by chain-transfer, as mentioned above. Thus the two are intimately entwined.
However, by using a PS seed latex with a low surface group concentration, adsorbing the surface-active SMDSo onto it, and then doing a second stage polymerization, we should be able to regulate the concentration of chemically bound surface functional groups. The seed latex was prepared using a silver ammine/sodium sulfite redox initiator that produces only •SO3– free-radical ions, which initiate polymer chains that thereby contain a sulfonate end-group. No emulsifier was used. The product PS colloid had a bimodal particle size distribution, with the principal population having a diameter of 700 nm; these were accompanied by some very small sizes. The latter were removed by fractional sedimentation at 1700 rpm, decantation of the smaller fraction, and ultrasonic redispersion of the large particles. This was repeated four times to give a product that showed no small sized particles under the transmission electron microscope (TEM). This colloid was subsequently purified by mixed bed ion exchange, which leads to all the surface sulfonate groups having H+ counterions. These, upon titration with aqueous Ba(OH)2, gave a surface charge density, σ0, of 0.34 μC cm–2.
The second stage, seeded emulsion polymerization, was undertaken according to the scheme below:
- • (1) adsorption of SMDSo onto PS Seed Colloid (deoxygenation of seed latex, addition of SMDSo solution, system maintained under nitrogen);
- • (2) monomer addition (addition of styrene, stirring until swelling equilibrium, or complete imbibing of styrene);
- • (3) emulsion polymerization (initiator – AIBN in dichloroethane – addition, polymerization at 55 °C);
- • (4) final work-up of the latex (solids content determined; mixed bed-ion exchange; solid content determined again, conductometric titration, TEM particle size determination).
A solution of SMDSo was added to the seed colloid in an amount calculated to give a final surface coverage of 185 Å2 per molecule. This was based on an adsorption isotherm of SMDSo on PS which was admittedly approximate, but which gave, with corrections, a surface saturation value of 31 Å2 per molecule. This compares to the comparable, generally accepted value for SDS on styrene of 45 Å2. The calculation was not crucial since some of the transurf would probably not be involved in chain initiation, some would form by-products, and the adsorption equilibrium would shift as the SMDSo was consumed.
In this experiment, azobis-iso-butyronitrile, AIBN, was used as the polymerization initiator because of its hydrophobic nature (it would concentrate in the particles thereby reducing the probability of new particle formation), and because it was not expected to oxidize the SMDSo (as would the very commonly used persulfate). All operations were carried out under O2-free nitrogen purging. An aqueous solution of the transurf was pipetted into the reaction flask containing the seed latex, and allowed to reach equilibrium adsorption under stirring for 45 min. Styrene monomer was added, and the mixture was stirred overnight to ensure complete absorption. Fifty microliters of 0.0064 M AIBN solution in 1,2-dichloroethane was added and stirred for another 3 h, time enough to totally diffuse into the polymer/monomer particles [9]. Polymerization was then carried out at 55 °C for 18 h. The results are summarized in Table 3.
Results of seeded emulsion polymerization
| PS Colloid | σ0 (μC cm–2) | Particle diameter (nm) | Å2 (sulfonate) |
| Seed | 0.34 | 700 | 4700 |
| Final | 5.3 | 740 | 300 |
| Theoretical final | 8.7 | 747 | 185 |
Particle sizes were estimated by Higher Order Tyndall Spectra (HOTS), which is useful for monodisperse colloids [10]. The final colloid, under careful TEM examination, showed no new particle formation. The surface coverage/surface charge density values were obtained as before after mixed bed ion exchange with highly purified resins [11] followed by titration with Ba(OH)2 reagent solution. This procedure produced a dramatic 16-fold increase in the surface functional group density, not as high as the theoretical value, no doubt because of the other fates to which this transurf is subject, as mentioned above.
3 Experimental section
3.1 Synthesis of sodium 10-bromo-1-decane sulfonate
A 3-l, round bottom flask containing a magnetic stirring bar was charged with 1660 ml ethanol (95%) and 200 g 1,10-dibromodecane (Aldrich, 97%). The dibromodecane was dissolved, and a solution containing 56.0 g of Na2SO3 in 660 ml of distilled water was added to the flask. The contents were heated under reflux with stirring for 30 h. The flask was cooled to room temperature, followed by cooling at 0 °C. The contents were decanted from the excess solid dibromodecane. The reaction mixture was evaporated to dryness at 60 °C on a rotary evaporator. The resulting solid was washed with petroleum ether (boiling range 30–60 °C). Further drying of the solid, to remove trace amounts of water, was often required to facilitate petroleum ether washings. Washings were continued until the dry solid was powdery in appearance. This indicated complete removal of the last traces of dibromodecane. The solid was washed with ice-cold acetone to remove the color. The product, sodium 10-bromo-1-decanesulfonate, was extracted from the dry solid with ethanol (100%), leaving a residue of NaBr.
3.2 Synthesis of 10-S-thiouronium-1-decanesulfonate (10-S-T)
Both 18 g of sodium 10-bromodecanesulfonate and 4.8 g of thiourea (Aldrich 98%) were dissolved in 150 ml water. The solution was heated under gentle reflux for one hour. After cooling the flask in ice, the resulting crystals were isolated by filtration, and washed several times with cold water. The 10-S-T could be recrystallized from 50% ethanol/water but it did not have a sharp melting point. After one recrystallization, 13.1 g (79% of theory) of purified product was obtained.
3.3 Guanidinium 10-mercapto-1-decanesulfonate (GMDSo)
Thirteen grams of the 10-S-thiuroniumdecanesulfonate and 400 ml of concentrated aqueous ammonia were placed in a 1-l round-bottom flask containing a magnetic stirring bar, and fitted with a reflux condenser. The contents were stirred and slowly warmed to 85 °C. Two 100-ml portions of concentrated aqueous ammonia were added over a period of about one half hour. After the 10-S-T had dissolved, the solution was stirred for 10 min. The stoppered flask was then cooled to room temperature and held there for 2 h.
The flask was placed on a rotary evaporator at 35 °C to remove ammonia and water. The product, GMDSo, was waxy in appearance, and water-soluble. In some instances, a white, water-insoluble precipitate formed as the ammonia was being removed. When this occurred, the yield of GMDSo could be improved by evaporating the mixture to dryness and redissolving the solid in 200 ml of aqueous ammonia at 85 °C. Rotary evaporation at 35 °C was then performed.
3.4 Sodium 10-mercapto-1-decanesulfonate (SMDSo)
A chromatographic column (63 cm × 2.5 cm diameter) was filled with 250 ml (wet volume) of cation exchange resin in its acid form. The resin had been cleaned previously by washing in 3 N NaOH, 3 N HCl and hot distilled water repeatedly, in that sequence up to five times. The GMDSo from the previous step was dissolved in 150 ml of deoxygenated, distilled water and filtered under nitrogen to remove any insoluble impurities. The solution was added under nitrogen to the top of the column, and the product was collected at the bottom. The resin was rinsed with several portions of distilled, deionized water. The combined eluates were brought to pH 7.0 with 1 N NaOH solution. The neutralized solution was evaporated to dryness, and the product, SMDSo, was extracted from its disulfide impurity with absolute ethanol. The product was recrystallized under nitrogen from absolute ethanol. Maximum recovery was achieved by cooling to about –10 °C in an ice-salt bath before filtration. After three recrystallizations, 3.25 g SMDSo were obtained, for an overall yield based on GMDSo of 27%. The melting point was 185.5–186.5 °C with decomposition.
3.5 Characterization of SMDSo
3.5.1 Qualitative tests for thiols
- (1) Lead acetate test: a few drops of saturated, ethanolic lead acetate were added to a sample of the unknown; a bright yellow precipitate of lead mercaptide was positive for this thiol. The test is negative for disulfide.
- (2) Nitroprusside test [12]: a 1.0% solution of sodium nitroprusside (Na2Fe(CN)5NO
3.5.2 Quantitative analysis
Iodine titration. The method of Harnish using ethanolic iodine as an oxidizing reagent was used [13]. Pyridine neutralizes the HI formed and catalyzes the reaction:
About 0.1 g of SMDSo was weighed accurately into a 50 ml volumetric flask, dissolved in 95% ethanol and diluted to the mark. Six drops of pyridine was added to a 20.0 ml aliquot of this solution, which was then titrated to the first faint, permanent I2 color.
3.5.3 Thin layer chromatography
Pre-coated TLC sheets (EM Reagents. Silica gel 60 F-254, 0.2 mm thickness) and a developing solution of 1-propanol/concentrated aqueous NH3 (70:30 by volume) were used for separation of the sulfonates. Detection of spots took place in an I2 vapor chamber. A 1–2-mm spot of a 5% solution of the sample was applied to the TLC plate, dried at 90 °C for several minutes, and then developed. They were then air-dried prior to placing in the developing chamber for 15 min.
3.5.4 Conductometric titration
The CMCs of both SDS and SMDSo were determined with a Mysels-type conductivity cell (Beckman Instruments) with a magnetic stirring bar along with an Industrial Instruments conductivity bridge. Measurements were made at 20 °C under O2-free N2. Sample solutions were initially at a concentration of about 2 × the estimated CMC. Serial dilutions were then made with distilled water. Only relative conductances were measured.
3.5.5 Surface tension measurements
A Rosano surface tensiometer with a Pt plate was used. The plate was cleaned with ethanol and distilled water, followed by flaming to red heat. Surface tension measurements of the SMDSo were made under water-saturated, O2-free N2 to avoid evaporation and oxidation to the highly surface-active disulfide. Deoxygenated water was used for serial dilutions. The cell was enclosed except for a nitrogen inlet and a glass tube through which the thread holding the Pt plate was suspended. After dilutions the solutions were stirred with an in situ magnetic bar, allowed to come to rest before the next measurement. Two sequential readings had to agree within 0.04 mN m–1 before they were considered reliable.
Adsorption isotherms at 25 °C were obtained by adding aliquots of a stock surfactant solution to a polystyrene latex, and making surface tension measurements after each addition.
3.6 Synthesis and characterization of polystyrene colloids
3.6.1 Biacetyl photoinitiation
Biacetyl (butanedione-2,3), distilled at atmospheric pressure, was used to make a 0.023 M aqueous stock solution, which was stored under refrigeration. Styrene was distilled at 45 °C /20 torr and kept at –10 °C. A typical emulsion polymerization procedure follows.
A 250-ml, 3-neck flask fitted with an inlet tube, addition funnel and containing a magnetic stirring bar, was charged with water, styrene and surfactant. A given amount of the biacetyl stock solution was placed in the addition funnel. Deoxygenation was effected by evacuating the system to 15 torr and back-filling with N2 (Matheson O2-free). The purging procedure was repeated seven or eight times. The flask was placed in a constant temperature bath at 55 °C, the biacetyl solution was added, and a ‘Hi-intensity’ halogen lamp was directed at the flask from a distance of about 10 cm. A typical reaction ran for 18–20 h. A typical recipe contained 85 ml H2O, 5 ml styrene, 10 ml of 0.023 M biacetyl solution, and 0.034 g of SMDSo.
3.6.2 AIBN thermal initiation
The initiator, 4,4'-azobis(cyanovaleric acid), AIBN, was recrystallized twice from absolute ethanol. Stock solutions of it in distilled water were prepared at concentrations from 1 × 10–3 M to 5 × 10–5 M. A 250-ml, 3-neck flask with magnetic stirrer was charged with initiator solution, styrene and surfactant. The system was exhaustively evacuated and purged with nitrogen as above, and brought to 60 °C. The reaction was run under O2-free N2 at this temperature for 20 h.
3.6.3 Bonding of Transurf to seed particles
The seed latex consisted of polystyrene (PS) particles of 700 nm diameter. It had been made monodisperse by fractional sedimentation, and converted to the acid form with highly purified ion-exchange resin. Surface charge was determined by conductometric titration.
18.7 g of this latex at 4.20% solids was placed in a 100 ml, 3-neck flask fitted with a nitrogen inlet and magnetic stirring bar. After exhaustive deoxygentation, as above, 1.0 ml of a 0.43% aqueous, deoxygenated solution of SMDSo was added under nitrogen. This was brought to adsorption equilibrium by stirring for 45 min. 0.19 ml of styrene was added and the system was brought to swelling equilibrium by stirring overnight. Fifty microliters of a 6.4 × 10–3 M solution of AIBN in dichloroethane was then added under N2, and stirred for 3 h to allow complete transfer of the initiator to the particles. The polymerization was carried out at 55 °C with stirring for 18 h as described above under ‘AIBN thermal initiation’ conditions.
3.6.4 Purification of the colloids
The produced latexes were purified by shaking with mixed bed ion-exchange resins. Resins were conditioned prior to use by the method described by Vanderhoff et al. [15]. A five-fold excess of resin, based on estimated equivalents of ions present in the latex, was used for each exchange. At least two or three exchanges were performed, after each of which the used resin was removed by filtration through glass wool.
3.6.5 Conductometric titration
We used standardized 2.98 × 10–3 N Ba(OH)2 solution as titrant, and a Manostat Digipet Ultramicro burette. Conductivity was determined with a recording amplifier based on a design by Wu et al. [16]. A Mysels-type cell manufactured by Beckman was employed. Dissolved CO2 was removed by purging the latex with N2.
3.6.6 Particle size measurements
Higher-order Tyndall Spectra (HOTS). This technique can be used when particle diameters of PS latexes are between ca. 200 and 850 nm, and the size distribution is very narrow. A clean beaker containing extremely dilute PS colloid was placed in the beam of a collimated incandescent light source in a darkened room. The scattering angle of each red and green band was recorded. Particle diameter could be determined to within 3% accuracy using calibration plots calculated by Liang [17]. Whenever possible, one should confirm by electron microscopy that there is only a single population of particles of the measured size. For example, Rayleigh-scattering particles present would not affect the HOTS of a population of larger, monodisperse particles.
Electron microscopy: we used both transmission (TEM) and scanning (SEM) methods. For the TEM, latexes were sprayed with a nebulizer onto grids with a carbon support. Particles were then shadowed with Pt/C under very high vacuum conditions to ensure sharp shadow edges. For the SEM, latexes were sprayed into cover slips, and sputter-coated with gold/palladium alloy.
Vous devez vous connecter pour continuer.
S'authentifier