

1 Introduction
The enantioselective hydrophosphonylation of carbonyls (or phospho-aldol reaction; for a recent review of the phospho-aldol reaction, see [1]) is a crucial and highly valuable phospho-transfer reaction for the synthesis of α-functionalised (commonly amino or hydroxy) phosphonate esters (Scheme 1), see, for example, [2]. Such phosphorus compounds have been found to possess broad-based medicinal application as, for example, bio-phosphate mimics [3], transition state models [4], anti-biotic [5], anti-viral [6] and in terms of anti-tumour activity [7]. Complexes of various metals have been found to act as effective catalysts for the reaction, notable titanium and lanthanide systems have been most closely investigated in the laboratories of Shibasaki [8], Shibuya [9] and Spilling [10].
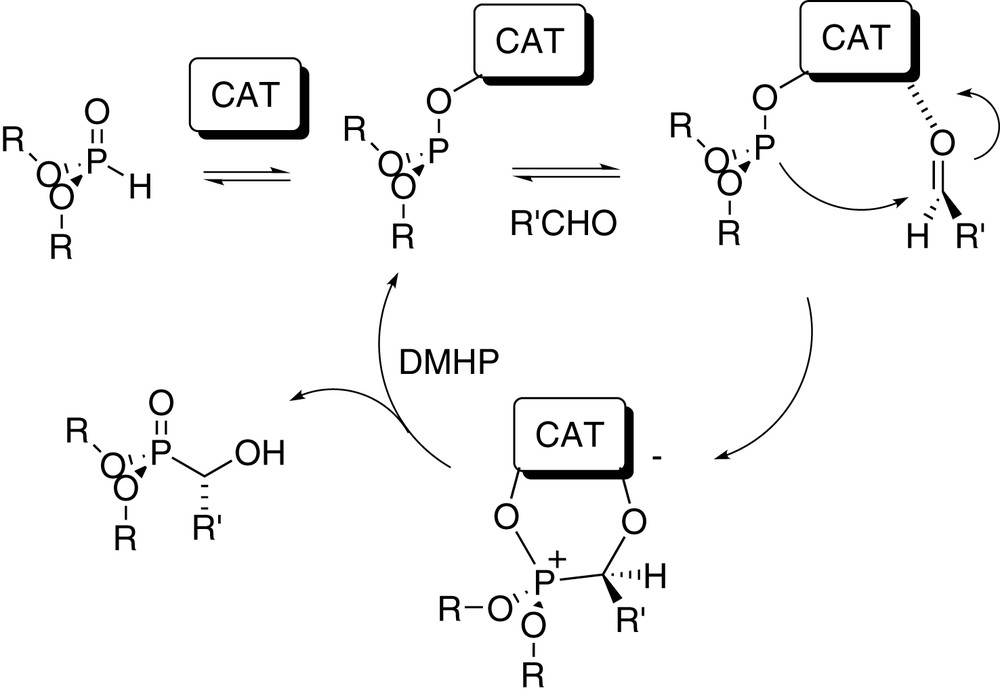
Catalytic cycle for the phospho-aldol reaction. R = alkyl, aryl etc.; CAT = catalyst.
In a series of recent publications we have explored the application of salcyen-metal complexes as catalysts for carbonyl hydrophosphonylation and recently reported that complexes of the general form [(R,R)-salcyen]AlX (X = Me, OSiMe2tBu) afford e.e.s < 50% in the addition of dimethyl-H-phosphonate (DMHP) to substituted benzaldehydes [11]. (In all our work, salcyen refers to the basic (R,R)-N,N′-bis(2'-hydroxybenzylidene)-trans-1,2-diaminocyclohexane framework as outlined in Scheme 2.) We encountered two rather unfortunate limitations with these salcyen complexes, apart from their rather modest stereoselectivities, which prompted us to search for new systems. We found salcyen complexes of aluminium to be both (i) quite slow catalysts and (ii) rather sensitive to quantities of water greater than that found under normal laboratory atmosphere conditions. Subsequently, we observed that certain aluminium complexes of tetrahydro-salcyen ligand frameworks (salcyans) were not only considerably more stable towards the presence of water but were also more active as phospho-aldol catalysts [12]. In this, our first full report on our salcyan work, we describe the first application of chiral salcyan complexes in asymmetric catalysis, specifically the phospho-aldol reaction and attempt to rationalize a most puzzling and, for us, exciting difference in stereochemical behaviour between salcyen and salcyan complexes with the same absolute stereochemistry of ligand framework.
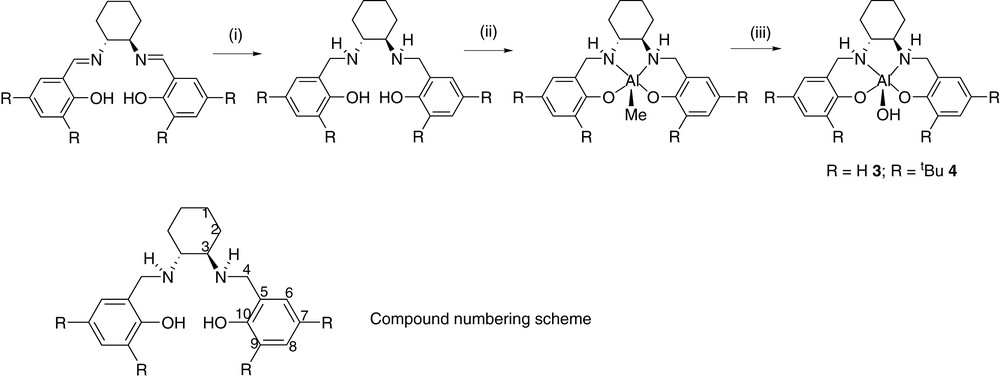
Syntheses of 3 and 4; (i) NaBH4; (ii) AlMe3; (iii) H2O. Compound numbering scheme also displayed.
Complexes [(R,R)-salcyan]Al(OH) 3 and [(R,R)-tBu4-salcyan]Al(OH) 4 were prepared using procedures similar to those developed by Atwood and co-workers for achiral derivatives [13]. We find that both 3 and 4 act as highly active precursors for the catalytic addition of dimethyl-H-phosphonate (DMHP) to benzaldehydes, but significant differences in catalytic behaviour to their close relatives, [(R,R)-salcyen]AlMe 5 and [(R,R)-tBu4-salcyen]AlMe 6 are noteworthy. In this contribution we describe full details of phospho-aldol catalysis via these (R,R)-salcyan systems, a comparison and rationalization, using molecular mechanics and semi-empirical analyses, of the differences in behaviour between (R,R)-salcyan and (R,R)-salcyen systems and reasoned arguments for the next generation of (R,R)-salcyan phospho-aldol catalysts.
2 Results and discussion
2.1 Syntheses and structures of [(R,R)-R4-salcyan]Al(OH) R = H 3, R = tBu 4
In Scheme 2 are outlined the individual steps in the syntheses of salcyan complexes 3 and 4. The initial stages of the procedures, preparing the ligand frameworks, have been reported on previously by ourselves and others and we see no reason to reproduce these here. Subsequent treatment of salcyans 1 and 2 with trimethylaluminium followed by exposure of the crude product to atmospheric moisture affords hydroxides 3 and 4 by analogy to achiral systems [13].
Crystals of compound 4, of suitable quality for single-crystal X-ray diffraction, have subsequently been obtained upon slow cooling of a pentane solution to –35 °C. Compound 4 is a hydroxy-bridged dimer with overall C2-symmetry [axis contains O(3) and O(4)] in which each aluminium atom is six-coordinate with a facial-type coordination of the salcyan ligand (Fig. 1 and Tables 1 and 2). Such an arrangement leads to each bridging oxygen atom being trans to either two oxygen atoms; O(3) being trans to O(36) and O(60) or trans to a nitrogen atom; O(4) being trans to N(5) and N(52). Within the primary coordination sphere of aluminium, distances between the metal and oxygen ligands of the salcyan ligands are 1.846(4) and 1.823(4) Å respectively for Al(1)–O(20) and Al(1)–O(36) and 1.832(4) and 1.854(4) Å for Al(2)–O(60) and Al(2)–O(76) respectively. These values are slightly longer than those for related distances in aluminium salcyen complexes (1.79–1.83 Å) although this may reflect the somewhat lower coordination geometries of the latter [11]. Expectedly longer are the distances between aluminium and the bridging hydroxyl oxygen atoms; although of perhaps more interest is the distinctly uneven nature of the two bridge units Al(1)–O(3)–Al(2) and Al(1)–O(4)–Al(2). The former angle, at 105.7(2) ° is some five degrees larger than that of the latter, a reflection of the shorter metal–oxygen distances of 1.871(4) Å and 1.882(4) Å respectively for Al(1)–O(3) and Al(2)–O(3) than for Al(1)–O(4) and Al(2)–O(4) of 1.945(4) Å and 1.934(4) Å. These distinctly asymmetrical structural parameters associated with the bridging ligands are undoubtedly linked to the associated trans-ligands (oxygen atoms for the shorter bridges and nitrogen for the longer) and result in quite different accessibilities with respect to potential reagents as clearly seen in a space-filling view of the bridge from the O(4) and O(3) directions (the RasMol generated images in Figs. 2a and b, respectively).
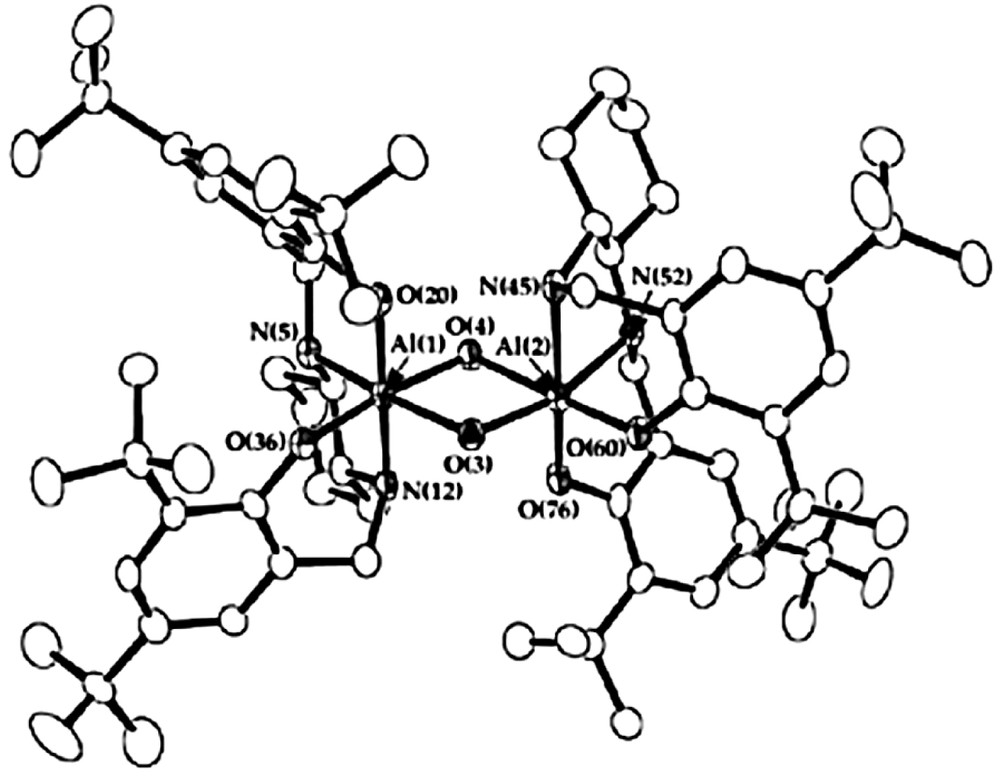
Molecular structure of 4.
Interatomic distances (Å) for 4 with e.s.d.s in parentheses
| Al1–O36 | 1.822(4) | Al1–O3 | 1.871(2) |
| Al1–O20 | 1.845(4) | Al1–N12 | 2.083(5) |
| Al1–O4 | 1.945(2) | Al1–Al2 | 2.990(2) |
| Al1–N5 | 2.090(5) | Al2–O76 | 1.855(4) |
| Al2–O60 | 1.832(4) | Al2–O4 | 1.9341 |
| Al2–O3 | 1.8810 | Al2–N52 | 2.103(5) |
| Al2–N45 | 2.098(5) | N5–C13 | 1.507(8) |
| N5–C6 | 1.484(8) | C6–C7 | 1.541(8) |
| C6–C11 | 1.537(9) | C8–C9 | 1.523(11) |
| C7–C8 | 1.514(10) | C10–C11 | 1.528(9) |
| C9–C10 | 1.538(9) | N12–C29 | 1.485(8) |
| C11–N12 | 1.481(7) | C14–C19 | 1.394(9) |
| C13–C14 | 1.496(9) | C15–O20 | 1.364(7) |
| C14–C15 | 1.407(8) | C16–C17 | 1.401(9) |
| C15–C16 | 1.399(9) | C17–C18 | 1.379(9) |
| C16–C21 | 1.548(9) | C18–C25 | 1.572(9) |
| C18–C19 | 1.368(9) | C21–C24 | 1.533(9) |
| C21–C23 | 1.528(10) | C25–C27 | 1.484(10) |
| C21–C22 | 1.541(10) | C25–C26 | 1.537(11) |
| C25–C28 | 1.526(10) | C30–C35 | 1.395(9) |
| C29–C30 | 1.498(8) | C31–O36 | 1.365(7) |
| C30–C31 | 1.422(9) | C32–C33 | 1.375(9) |
| C31–C32 | 1.418(8) | C33–C34 | 1.378(10) |
| C32–C37 | 1.542(10) | C34–C41 | 1.540(9) |
| C34–C35 | 1.386(9) | C37–C39 | 1.533(10) |
| C37–C38 | 1.511(10) | C41–C42 | 1.492(12) |
| C37–C40 | 1.544(9) | C41–C44 | 1.538(12) |
| C41–C43 | 1.514(11) | N45–C53 | 1.485(8) |
| N45–C46 | 1.484(8) | C46–C51 | 1.519(9) |
| C46–C47 | 1.517(9) | C48–C49 | 1.529(11) |
| C47–C48 | 1.566(10) | C50–C51 | 1.523(9) |
| C49–C50 | 1.544(10) | N52–C69 | 1.487(8) |
| C51–N52 | 1.470(8) |
Angles between interatomic vectors (°) for 4. E.s.d.s in parentheses
| O36–Al1–O20 | 96.32(19) | O36–Al1–O3 | 99.38(15) |
| O20–Al1–O3 | 95.09(14) | O36–Al1–O4 | 173.16(17) |
| O20–Al1–O4 | 89.66(15) | O3–Al1–O4 | 76.71(7) |
| O36–Al1–N12 | 90.2(2) | O20–Al1–N12 | 170.0(2) |
| O3–Al1–N12 | 91.34(15) | O4–Al1–N12 | 84.34(15) |
| O36–Al1–N5 | 93.71(19) | O20–Al1–N5 | 92.0(2) |
| O3–Al1–N5 | 164.33(16) | O4–Al1–N5 | 89.38(16) |
| N12–Al1–N5 | 80.0(2) | O36–Al1–Al2 | 136.42(15) |
| O20–Al1–Al2 | 93.03(13) | O3–Al1–Al2 | 37.27(4) |
| O4–Al1–Al2 | 39.44(4) | N12–Al1–Al2 | 87.44(14) |
| N5–Al1–Al2 | 128.46(15) | O60–Al2–O76 | 97.60(19) |
| O60–Al2–O3 | 98.06(13) | O76–Al2–O3 | 95.63(13) |
| O60–Al2–O4 | 172.09(13) | O76–Al2–O4 | 88.91(13) |
| O3–Al2–O4 | 76.7 | O60–Al2–N45 | 91.50(19) |
| O76–Al2–N45 | 167.9(2) | O3–Al2–N45 | 90.97(14) |
| O4–Al2–N45 | 82.71(13) | O60–Al2–N52 | 93.96(19) |
| O76–Al2–N52 | 91.60(19) | O3–Al2–N52 | 165.01(14) |
| O4–Al2–N52 | 90.32(14) | N45–Al2–N52 | 79.76(19) |
| O60–Al2–Al1 | 134.82(15) | O76–Al2–Al1 | 93.10(12) |
| O3–Al2–Al1 | 37.04(4) | O4–Al2–Al1 | 39.70(4) |
| N45–Al2–Al1 | 86.06(14) | N52–Al2–Al1 | 129.59(15) |
| Al1–O3–Al2 | 105.68(7) | Al2–O4–Al1 | 100.86(6) |
| C6–N5–C13 | 112.8(5) | C6–N5–Al1 | 109.5(4) |
| C13–N5–Al1 | 110.4(3) | N5–C6–C11 | 106.5(5) |
| N5–C6–C7 | 116.5(5) | C11–C6–C7 | 108.9(5) |
| C8–C7–C6 | 110.0(6) | C7–C8–C9 | 111.6(6) |
| C8–C9–C10 | 111.8(6) | C11–C10–C9 | 109.8(5) |
| N12–C11–C10 | 114.5(5) | N12–C11–C6 | 109.0(5) |
| C10–C11–C6 | 110.5(5) | C11–N12–C29 | 110.8(4) |
| C11–N12–Al1 | 113.8(4) | C29–N12–Al1 | 112.0(4) |
| C14–C13–N5 | 110.5(5) | C19–C14–C15 | 120.6(5) |
| C19–C14–C13 | 120.2(5) | C15–C14–C13 | 119.1(5) |
| O20–C15–C16 | 122.6(5) | O20–C15–C14 | 118.0(5) |
| C15–O20–Al1 | 126.0(3) | N12–C29–C30 | 111.0(5) |
| C35–C30–C31 | 121.0(5) | C35–C30–C29 | 120.0(6) |
| C31–C30–C29 | 119.0(5) | O36–C31–C32 | 123.0(6) |
| O36–C31–C30 | 118.9(5) | C33–C34–C41 | 121.0(6) |
| C35–C34–C41 | 122.5(7) | C34–C35–C30 | 120.8(7) |
| C31–O36–Al1 | 133.1(4) | C46–N45–C53 | 111.9(4) |
| C46–N45–Al2 | 113.1(4) | C53–N45–Al2 | 111.6(4) |
| N45–C46–C47 | 113.6(5) | N45–C46–C51 | 111.0(5) |
| N52–C51–C46 | 108.1(5) | N52–C51–C50 | 116.5(5) |
| C46–C51–C50 | 109.5(5) | C51–N52–C69 | 113.8(5) |
| C51–N52–Al2 | 109.3(3) | C69–N52–Al2 | 111.0(4) |
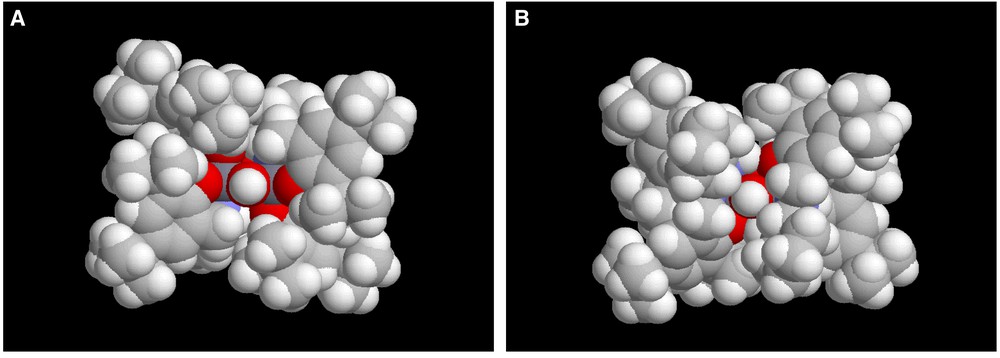
Structure of 4 in space-filling mode, emphasising (a) the more open bridge face of O(4); (b) the less open bridge face of O(3) (oxygen atoms shown in red).
Room temperature solution phase NMR analyses are consistent with 4 adopting a similar structure to that displayed in the solid phase. Consistently, each of the tert-butyl groups in the salcyan ligand framework occupies distinct chemical environments in the solid state. Whilst it is also true that each tert-butyl group would be expected to be distinct even in a monomeric form of 4, electrospray mass spectrometry in THF/MeOH solvent affords both dimeric fragments at m/z 1168 assigned to {[Al]–O–[Al]}+ and m/z 608 due to the monomeric metabolite [Al]–OMe. In pure MeOH, dimeric and monomeric fragments resulting from reaction with solvent are most clearly observed including; m/z 1212 {[Al]–OMe}2+, 615 [Al]–OHNa+, 629 [Al]–OMeNa+.
2.2 [(R,R)-R4-salcyan]AlX and [(R,R)-R4-salcyen]AlX complexes as phospho-aldol catalysts
We explore below the potential of (R,R)-R4-salcyan complexes 3 and 4 as catalytic pre-cursors for the phospho-aldol reaction and perform SAR analyses in comparison to structurally related (R,R)-R4-salcyen systems.
In Table 3 are collected the results of phospho-aldol catalysis reactions on various 4-substituted benzaldehydes under identical conditions, when catalysed by complex 4 and its salcyen cousins [(R,R)-salcyen]AlMe 5 and [(R,R)-tBu4-salcyen]AlMe 6 [11]. We have not included enantioselectivity data for salcyan complex 3 since for each substituted benzaldehyde the e.e. returned as <10% and were found to be rather difficult to reproduce, we believe due to questions over compound purification (vide infra). From the data in Table 3, it is immediately obvious from the figures, both (R,R)-salcyen and (R,R)-R4-salcyan systems have some rather extraordinary differences that, we feel, serve to highlight important structural features. We have shown previously that with [(R,R)-Rn-salcyen]AlX catalyst precursors, the nature of the X function, whilst influencing activity, does not unduly influence enantioselectivity of product. [11] Therefore, we envisage that the change in absolute configuration of α-hydroxyphosphonate ester, (MeO)2P(=O)CH(OH)(C6H4–R) from R to S when catalyst precursor changes from 5 to 4 reflects the different structures at the metal centre that result from the change in ligand structure from salcyen to salcyan. Particularly intriguing are the twin facts that; (i) within the salcyen system, the (R,R)-H4-salcyen framework promotes a greater enantioselectivity than (R,R)-tBu4-salcyen, whilst the reverse trend is true for the salcyan systems and (ii) changing from an (R,R)-R4-salcyen ligand framework to an (R,R)-R4-salcyan framework can lead to changes in enantioselectivity, Δe.e. of >100!
Phospho-aldol addition DMHP (dimethyl-H-phosphonate) to aldehydes in THF solvent in the presence of catalytic precursor complexes 4 and [(R,R)-salcyen]AlMe 5 and [(R,R)-tBu4-salcyen]AlMe 6. All reactions were performed in air at 298 K, using a catalyst loading of 5 mol% for the salcyen complexes and 1 mol% for the salcyan complexes and reaction time of 4 h unless specified otherwise. Catalyst loadings 4 are calculated as mole % of the dimer
| Run | E.E. (config.) Complex 4 | E.E. (config.) Complex 5 | E.E. (config.) Complex 6 | 4-XC6H4CHO |
| 1 | 59 (S) | 41 (R) | 3 (R) | X = H |
| 2 | 42 (S) | 24 (R) | 13 (S) | X = Br |
| 3 | 57 (S) | 49 (R) | 3 (S) | X = Me |
| 4 | 47 (S) | 46 (R) | — | X = OMe |
| 5 | 18 (S) | 10 (R) | — | X = NO2 |
| 6 | 22 (S) | — | — | X = NMe2 |
What are the causes for such a change in stereoselectivity? We believe that even though both salcyen and salcyan ligands possess the same absolute configuration of backbone, their structural properties are quite different, principally as a result of the change in hybridisation from N=CHR in salcyen to NHCH2R in the salcyan system. A single-crystal X-ray analysis of (R,R)-tBu4-salcyan 2 has been performed and the molecular structure illustrated in Fig. 3 (representative bond distances and angles in Tables 6 and 7). From Fig. 3, there is a clear structural case for the salcyan systems to prefer trans-periplanar arrangements of CH and NH functionalities as illustrated in the H–C(2)–N(2)–H dihedral. That this conformation is not shared in the corresponding H-C(1)–N(1)–H dihedral is, we believe, a result of intramolecular hydrogen bonding between O(16)–H(16) and N(2) and between O(9)–H(9) and N(1) (Table 8).
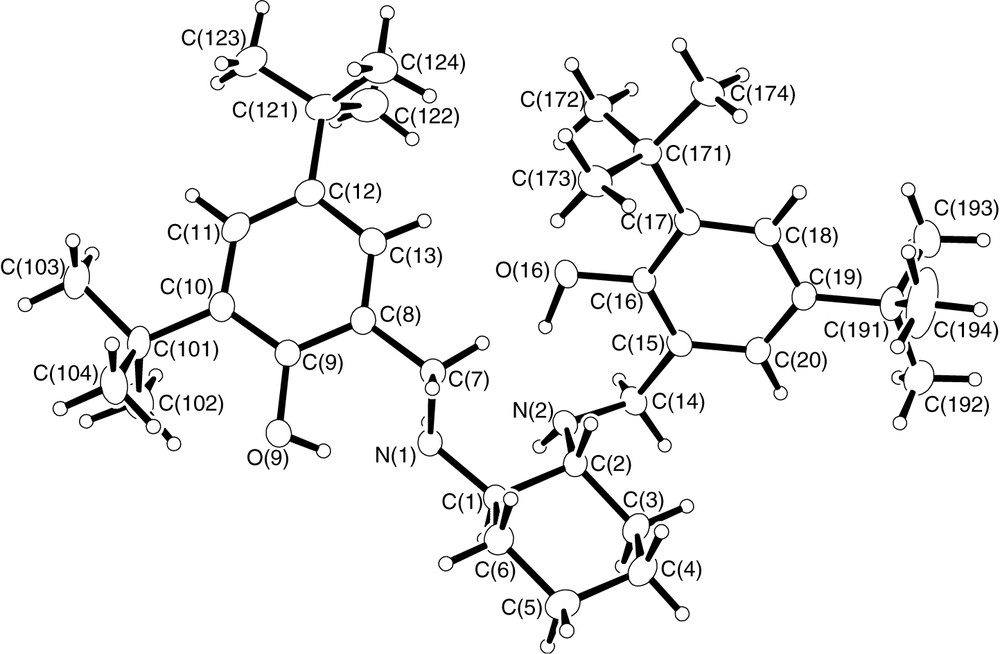
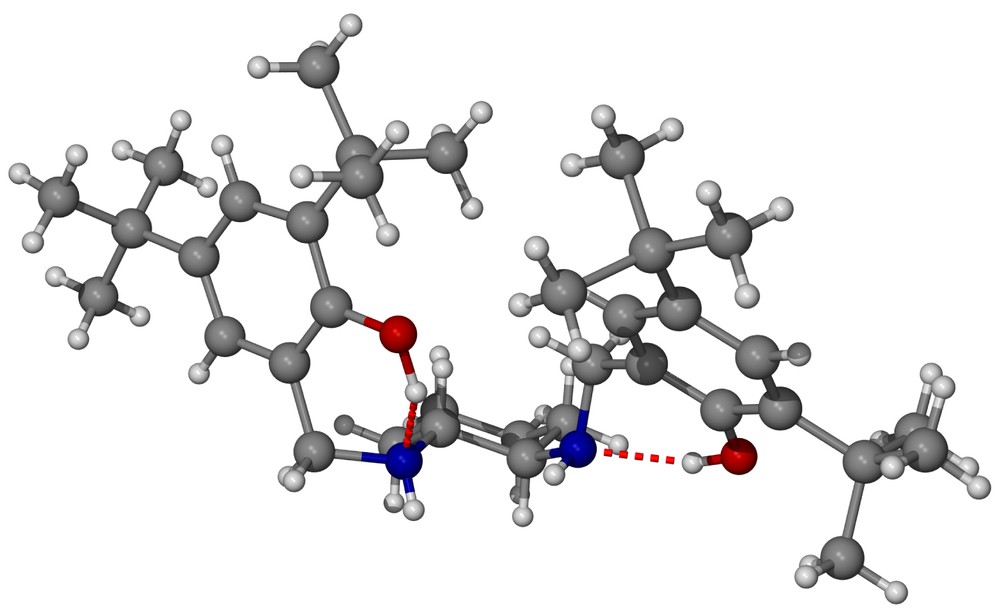
(a) Molecular structure of (R,R)-tBu4-salcyan 2; (b) molecular structure of (R,R)-tBu4-salcyan 2 emphasising chair structure of the cyclohexane ring and trans-periplanar arrangement of H–C(2)–N(2)–H.
Interatomic distances (Å) for 2 with e.s.d.s in parentheses
| C(1)–N(1) | 1.476(3) | C(1)–C(6) | 1.522(3) |
| C(1)–C(2) | 1.550(3) | C(2)–N(2) | 1.474(3) |
| C(2)–C(3) | 1.533(3) | C(3)–C(4) | 1.528(4) |
| C(4)–C(5) | 1.513(4) | C(5)–C(6) | 1.524(4) |
| N(1)–C(7) | 1.479(3) | C(7)–C(8) | 1.514(3) |
| C(8)–C(13) | 1.379(3) | C(8)–C(9) | 1.398(3) |
| C(9)–O(9) | 1.378(3) | C(9)–C(10) | 1.400(3) |
| C(10)–C(11) | 1.396(4) | C(10)–C(101) | 1.541(3) |
| C(101)–C(102) | 1.535(5) | C(101)–C(104) | 1.543(5) |
| C(101)–C(103) | 1.550(4) | C(11)–C(12) | 1.399(4) |
| C(12)–C(13) | 1.393(3) | C(12)–C(121) | 1.530(4) |
| C(121)–C(123) | 1.492(5) | C(121)–C(124) | 1.561(5) |
| C(121)–C(122) | 1.566(5) | N(2)–C(14) | 1.479(3) |
| C(14)–C(15) | 1.508(3) | C(15)–C(20) | 1.391(3) |
| C(15)–C(16) | 1.398(3) | C(16)–O(16) | 1.373(3) |
| C(16)–C(17) | 1.414(3) | C(17)–C(18) | 1.397(3) |
| C(17)–C(171) | 1.531(3) | C(171)–C(172) | 1.540(3) |
| C(171)–C(173) | 1.542(3) | C(171)–C(174) | 1.544(3) |
| C(18)–C(19) | 1.407(3) | C(19)–C(20) | 1.387(3) |
| C(19)–C(191) | 1.530(3) | C(191)–C(194) | 1.499(4) |
| C(191)–C(193) | 1.538(4) | C(191)–C(192) | 1.555(4) |
Angles between interatomic vectors (°) for 2. E.s.d.s in parentheses
| N(1)–C(1)–C(6) | 111.01(19) | N(1)–C(1)–C(2) | 115.17(18) |
| C(6)–C(1)–C(2) | 111.02(18) | N(2)–C(2)–C(3) | 116.02(19) |
| N(2)–C(2)–C(1) | 108.15(17) | C(3)–C(2)–C(1) | 109.13(19) |
| C(4)–C(3)–C(2) | 111.3(2) | C(5)–C(4)–C(3) | 111.3(2) |
| C(4)–C(5)–C(6) | 110.8(2) | C(1)–C(6)–C(5) | 111.5(2) |
| C(1)–N(1)–C(7) | 116.66(17) | N(1)–C(7)–C(8) | 109.66(18) |
| C(13)–C(8)–C(9) | 120.0(2) | C(13)–C(8)–C(7) | 120.9(2) |
| C(9)–C(8)–C(7) | 119.1(2) | O(9)–C(9)–C(8) | 119.3(2) |
| O(9)–C(9)–C(10) | 119.8(2) | C(8)–C(9)–C(10) | 120.8(2) |
| C(11)–C(10)–C(9) | 116.5(2) | C(11)–C(10)–C(101) | 122.5(2) |
| C(9)–C(10)–C(101) | 121.0(2) | C(102)–C(101)–C(10) | 109.4(2) |
| C(102)–C(101)–C(104) | 110.1(3) | C(10)–C(101)–C(104) | 110.3(3) |
| C(102)–C(101)–C(103) | 107.6(3) | C(10)–C(101)–C(103) | 111.4(2) |
| C(104)–C(101)–C(103) | 108.0(3) | C(10)–C(11)–C(12) | 124.6(2) |
| C(13)–C(12)–C(11) | 116.1(2) | C(13)–C(12)–C(121) | 120.2(2) |
| C(11)–C(12)–C(121) | 123.6(2) | C(123)–C(121)–C(12) | 112.6(3) |
| C(123)–C(121)–C(124) | 107.9(4) | C(12)–C(121)–C(124) | 109.7(3) |
| C(123)–C(121)–C(122) | 112.4(4) | C(12)–C(121)–C(122) | 108.8(2) |
| C(124)–C(121)–C(122) | 105.0(3) | C(8)–C(13)–C(12) | 121.9(2) |
| C(2)–N(2)–C(14) | 116.91(18) | N(2)–C(14)–C(15) | 111.93(18) |
| C(20)–C(15)–C(16) | 119.9(2) | C(20)–C(15)–C(14) | 119.9(2) |
| C(16)–C(15)–C(14) | 120.2(2) | O(16)–C(16)–C(15) | 119.14(19) |
| O(16)–C(16)–C(17) | 119.62(19) | C(15)–C(16)–C(17) | 121.2(2) |
| C(18)–C(17)–C(16) | 116.2(2) | C(18)–C(17)–C(171) | 122.37(19) |
| C(16)–C(17)–C(171) | 121.37(19) | C(17)–C(171)–C(172) | 111.36(18) |
| C(17)–C(171)–C(173) | 108.86(18) | C(172)–C(171)–C(173) | 110.0(2) |
| C(17)–C(171)–C(174) | 112.2(2) | C(172)–C(171)–C(174) | 106.72(19) |
| C(173)–C(171)–C(174) | 107.61(19) | C(17)–C(18)–C(19) | 124.1(2) |
| C(20)–C(19)–C(18) | 117.2(2) | C(20)–C(19)–C(191) | 122.3(2) |
| C(18)–C(19)–C(191) | 120.5(2) | C(194)–C(191)–C(19) | 109.2(2) |
| C(194)–C(191)–C(193) | 111.3(3) | C(19)–C(191)–C(193) | 109.7(2) |
| C(194)–C(191)–C(192) | 109.6(3) | C(19)–C(191)–C(192) | 112.0(2) |
| C(193)–C(191)–C(192) | 105.0(2) | C(19)–C(20)–C(15) | 121.5(2) |
Hydrogen-bonded distances (Å) and angles (°) in 2. Standard uncertainties are included in parentheses for values that do not involve constrained hydrogen atoms
| Atoms (D–HA) | D–H | HA | DA | ∠DHA |
| O(16)–H(16)N(2) | 1.04(4) | 1.76(4) | 2.685(3) | 146.(3) |
| O(9)–H(9)N(1) | 0.83(4) | 1.91(4) | 2.685(3) | 155.(3) |
The most interesting observation though is that once (R,R)-tBu4-salcyan 2 becomes coordinated to aluminium in complex 4, both H–C–N–H dihedrals become trans-periplanar. This directional feature, coupled with the increased flexibility in moving from an sp2-hybridised function N=CHR in salcyen’s to a more conformationally flexible sp3-hybridised NHCH2R in 2, gives salcyan ligands a greater degree of coordination flexibility; a feature we return to below.
In addition to the differences between (R,R)-R4-salcyen and (R,R)-R4-salcyan complexes, there are clear differences in enantioselectivity (e.e.) associated with the electronic nature of the substituted benzaldehyde. Specifically, for both (R,R)-R4-salcyen and (R,R)-R4-salcyan complexes 4 and 5, e.e.s tend to be reduced in the presence of inductively electron withdrawing groups (Br, NO2) in Table 3. A trend appears somewhat more apparent within the salcyen system which we have commented upon previously and which we have taken to reflect carbonyl coordination to the metal centre being resulting in increased e.e. [11].
Solvent affects activity but not enantioselectivity. Upon changing solvent from CH2Cl2 through toluene to THF conversion (%) increases from 32% (CH2Cl2) to 75% (THF), a result, we believe, of the greater coordination ability of THF over the other two (vide infra). Enantioselectivities remain unchanged at 41(1)% over the three solvents. That solvent donor power has an influence over reactivity yet not apparently over enantioselectivity lead us to speculate that perhaps it is necessary for the {[(R,R)-tBu4-salcyan]Al(OH)}2 dimer to dissociate into a monomeric species before catalysis can be initiated. Results of phospho-aldol catalysis with differing loadings of complex 4 as pre-catalyst are also revealing in this connection (Table 4). Upon decreasing the catalyst load from 45 mol% (calculated as dimer) to 1 mol%, the conversion after 4 h remained excellent (>95%) but, most intriguingly, the e.e. increased from 18% (run 1) to 59% (run 5), in each case favouring the S-product enantiomer. Similar behaviour has been noted by North and Belekon in their study of carbonyl cyanation catalysed by (R,R)-salcyen complexes and explained on the basis that a more stereoselective complex results upon reaction of a catalytic precursor complex with adventitious water (see, for example, [14]). Given the differences between the North/Belekon system and our own, a modified explanation is necessary in our scenario. Increased dilution should result in increased dissociation of the dimeric pre-catalyst complex and formation of monomeric [(R,R)-tBu4-salcyan]Al(OH)(THF). Consequently, one possibility might be that reaction proceeds via two pathways, (i) monometallic and (ii) bimetallic. Perhaps the monomer is capable of bringing both substrate and reagent together at a single metal centre whilst in the dimer one metal centre may activate the phosphorus reagent whilst the other metal centre activates the carbonyl (Scheme 3a). A second possibility is that the reaction mechanism involves two different pathways, (i) one in which carbonyl binds to a metal centre prior to [P–C] bond formation (Scheme 1), and (ii) one where the carbonyl does not bind to a metal (Scheme 3b). The relative rates of these two processes will obviously be dependent upon the concentration of metal complex in solution. Studies are ongoing in order to address these mechanistic details.
Phospho-aldol addition of DMHP (dimethyl-H-phosphonate) to aldehydes in THF solvent in the presence of various pre-catalyst complex 4 as a function of catalyst loading. All reactions were performed in air at 298K unless specified otherwise
| Run | Cat. loada (%) | Conversion (%)a | E.E. (%)b |
| 1 | 45 | 100 | 18 (S) |
| 2 | 20 | 100 | 26 (S) |
| 3 | 10 | 100 | 39 (S) |
| 4 | 5 | 100 | 48 (S)c |
| 5 | 1 | 95 | 59 (S) |
| 6 | 0.5 | 74d | 58 (S) |
| 7 | 0.2 | 54e | 52 (S) |
a Catalyst loads refer to mole percentage of dimmer. Conversions (Con) obtained after 4hrs. No product other than α-hydroxyphosphonate ester was observed.
b Determined by 31P{1H}-NMR in the presence of quinine as a chiral solvating agent (±2%). Absolute configurations determined to be S by correlation of 31P-NMR shifts to optical rotations.
c Yield = 87% and e.e. = 53%(S) when reaction is performed at 273K.
d After 12 h.
e After 24 h.
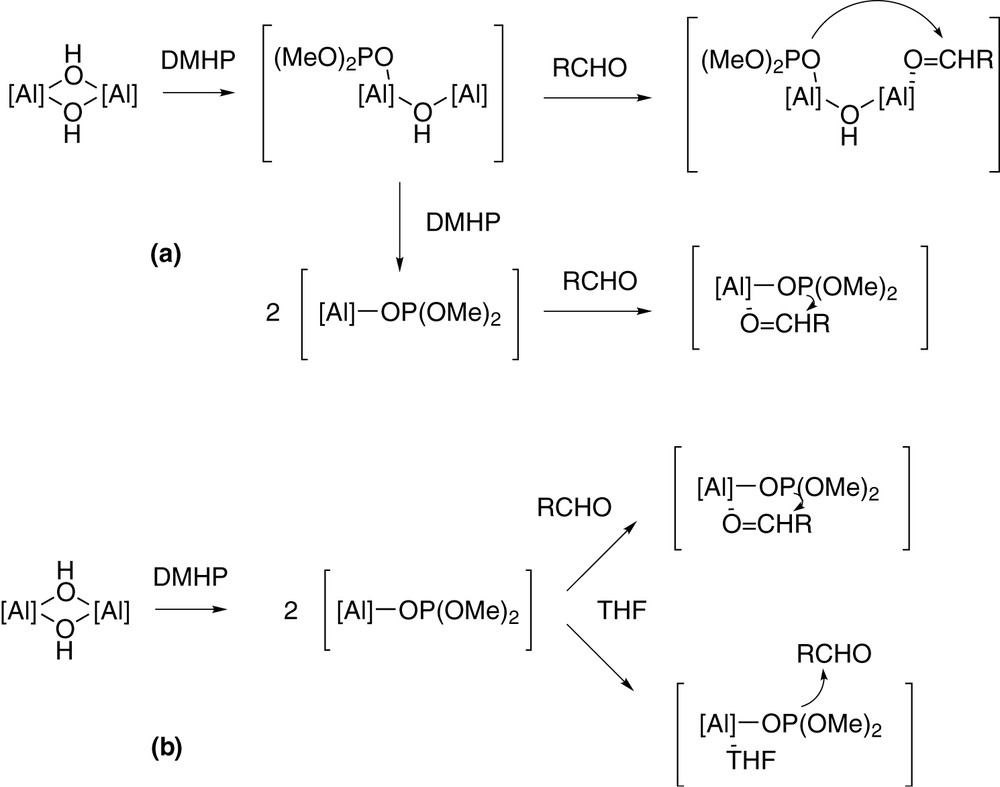
Possible mechanisms of monometallic and bimetallic pathways for the phospho-aldol process.
Finally, as reflected in Table 5, changing the diorgano-H-phosphonate from dimethyl to diethyl has negligible effect upon the enantioselectivity of reaction with 4-substituted benzaldehydes (Δe.e. ±1–7%) whilst reactivity decreases in that order. Presumably these two results indicate that the nature of the phosphorus alkyl substituent has relatively little influence over the stereochemically-controlling [P-C] bond forming step but that conversion of pre-catalyst to the important catalyst turnover species is less facile to some degree.
Phospho-aldol addition of DMHP and DEHP (diethyl-H-phosphonate) to aldehydes in THF solvent in the presence of pre-catalyst complex 4. All reactions were performed in air at 298 K over a reaction period of 4 h using 1 mol% dimer 4
| H-phosphonate | 4-XC6H4CHO | Conversion (%)a | E.E. (%)b |
| DMHP | X = H | 95 | 59 (S) |
| DMHP | X = Br | 75 | 42 (S) |
| DMHP | X = Me | 76 | 57 (S) |
| DMHP | X = OMe | 91 | 47 (S) |
| DMHP | X = NO2 | 64 | 18 (S) |
| DEHP | X = H | 50 | 61 (S) |
| DEHP | X = Br | 47 | 49 (S) |
| DEHP | X = Me | 52 | 56 (S) |
| DEHP | X = OMe | 69 | 48 (S) |
| DEHP | X = NO2 | 14 | 15 (S) |
a No product other than α-hydroxyphosphonate ester was observed.
b Determined by 31P{1H}-NMR in the presence of quinine as a chiral solvating agent (±2%). Absolute configurations determined to be S by correlation of 31P-NMR shifts to optical rotations.
2.3 Phospho-aldol catalysis via [(R,R)-tR4-salcyan] and [(R,R)-tR4-salcyen] complexes. Why should the same ligand backbone stereochemistry result in opposite product stereochemistry?
We are currently building a full picture of the mechanism(s) of phospho-aldol catalysis via a detailed structure-activity relationship (SAR) analysis. Whilst some of the results of this extensive study are reported here, we believe it is of significant interest to identify one key structural difference between (R,R)-R4-salcyen and (R,R)-R4-salcyan systems; the presence of a sterically active NH group within the latter. In Fig. 4, we illustrate the preferred geometry of orientation for an (R,R)-salcyan model system, a 1,3-diazacyclopentane ring system fused to a (R,R)-cyclohexane ring. Molecular mechanics calculations at the MM2 level (Chem3D) reveal that the trans form (a) in Fig. 4 is ca. 2 kJ mol–1 more stable than the cis form (b).
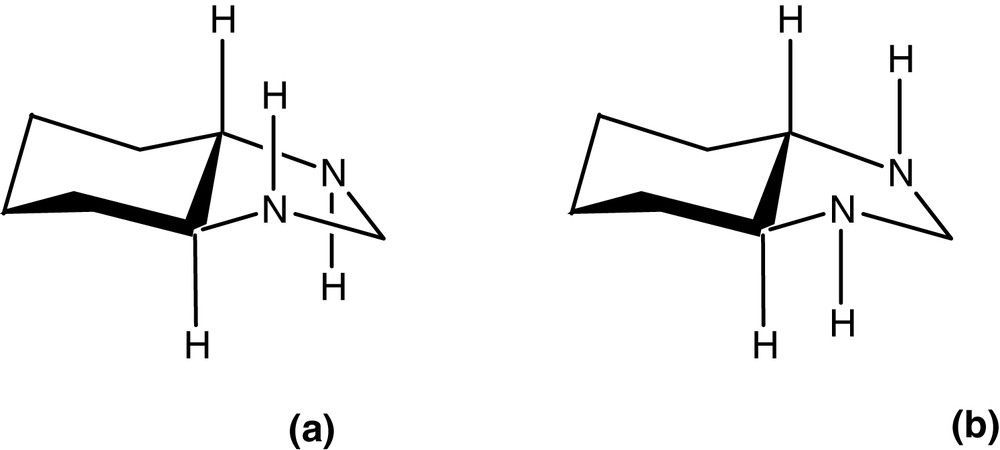
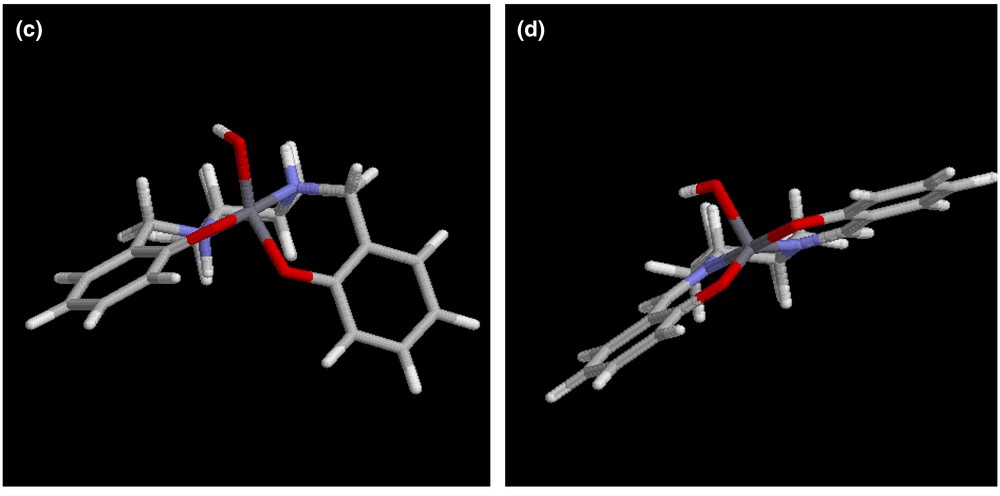
Anti-periplanar (a) and syn-periplanar (b) geometries of octahydrobenzoimidzole. MM+ molecular mechanics images (computed using HyperChem 7.0 and visualized with RasMol 2.7.1.1.) of [(R,R)-salcyen]Al(OH) (c) and [(R,R)-salcyan]Al(OH) (d) calculated using Chem3D.
We envisage that this steric preference within the salcyan system, albeit energetically small in itself, may be of significance in controlling the sense of ligand twisting in the metal coordination sphere. For instance, molecular mechanics calculations at the MM+ level on two related complexes; [(R,R)-salcyen]Al(OH) and [(R,R)-salcyan]Al(OH) reveal quite distinct differences in twisting at the metal centre (Fig. 4c and d). We envisage that a carbonyl substrate would approach [(R,R)-salcyen]Al(OH) along a vector approximately trans to the hydroxyl (or phosphito) group whereas we envisage a carbonyl preferring to approach [(R,R)-salcyan]Al(OH) along a vector trans to cyclohexyl nitrogen. It is not inconceivable that such disparate binding arrangements may in some, as yet undefined way, influence the absolute sense of stereo-induction in the phospho-aldol process. Comparison with X-ray structural data on related derivatives [(R,R)-salcyen]AlMe and {[(R,R)-tBu4-salcyan]Al(OH)}2 leads us to conclude that these structural differences translate from in silico to the solid state.
3 Conclusions
The enantioselective addition of diorgano-H-phosphonates to aldehydes, the phospho-aldol reaction, is catalysed in >60% e.e. by sub-1 mol% loadings of (R,R)-salcyan-Al complexes at room temperature under aerobic conditions; the first time that such catalysts have been made available. Absolute configurations are opposite to those observed when cousin (R,R)-salcyen-Al complexes are used as catalyst pre-cursors despite the two systems having the same absolute configuration of ligand framework! Synthetic and computational studies suggest that these differences may be connected to the structurally active NH function in (R,R)-salcyan systems. Structure-activity studies suggest that solvent and nature of the diorgano-H-phosphonate influence catalyst activity but not stereoselectivity whilst the electronic nature of the carbonyl substrate plays a role in influencing both, generally the most reactive carbonyls afford the lower enantioselectivities. Single-crystal X-ray analyses reveal that [(R,R)-tBu4-salcyan]Al(OH) exists in the solid state as a hydroxyl-bridged dimer which appears to translate also to solution phase. Elucidation of the precise nature of the catalytically active species is the subject of on-going study in this laboratory.
4 Experimental
4.1 General methods and techniques
All manipulations were carried out under aerobic conditions unless specified otherwise. Solvents used were pre-dried over sodium wire or calcium chloride before being refluxed over sodium (toluene), sodium and benzophenone (tetrahydrofuran), lithium aluminium hydride (pentane) and calcium hydride (dichloromethane); all solvents were then deoxygenated by purging with dinitrogen gas prior to use. Reagents such as cis/trans-cyclohexane-1,2-diamine, l-(+)-tartaric acid, lithium aluminium hydride, sodium borohydride, trimethylaluminium, triethylamine and selected aldehydes (unless stated otherwise) were purchased from commercial sources and used as received. Trimethylamine was distilled and stored over molecular sieves 4 Å under dinitrogen. Optical rotations are measured using an Optical Activity AA 10 polarimeter operating at 589.44 nm (sodium d-line). Microanalyses were performed in the Department of Chemistry Microanalysis Laboratory by Mr. M. Huscroft. Infrared spectra were recorded using KBr discs or Nujol mulls with a MIDAC FT-IR grating spectrophotometer (4000–600 cm–1). Ms. T. Marinko-Covell collected mass spectrometric data on a VG Autospec instrument (operating at 70 eV for electron impact). All NMR spectra were collected at 298K using a Bruker ARX 250 and Bruker ARC500 instruments (operating frequencies for 1H, 13C and 31P of 250.1, 500, 62.9, and 101.6 M Hz, respectively). Chemical shifts are referenced either to residual protons in the solvent or to tetramethylsilane for 1H and 13C and to 85% H3PO4 for 31P. Chemical shift values are given in ppm and assignments are according to the numbering profile of Scheme 2. (R,R)-Cyclohexane-1,2-diamine was resolved using the published procedure15 and was recrystallised to an [α]D of +12.5 (c = 4 g/dl, H2O) prepared as described previously. The compounds (R,R)-salcyen and (R,R)-tBu4-salcyen were prepared as described previously [15]. Molecular mechanics and semi-empirical analyses were performed at the MM+ and AM1 levels respectively using the HyperChem 7.0 package (HyperCube Inc., 1115 NW 4th St., Gainsville, Florida, USA) [(R,R)-N,N′-Bis(2′-hydroxy-substituted-benzylidene)-trans-1,2-diaminocyclohexane = substituted (R,R) salcyen.].
4.2 Synthesis of (R,R)-salcyan 1
(R,R)-Salcyen (2.5 g, 7.8 mmol) was dissolved in methanol (25 cm3) under an atmosphere of dinitrogen. The solution was heated to 60 °C whereupon NaBH4 (1.7 g, 44.7 mmol) was added carefully in small portions after which the combined mixture was heated to reflux for a period of 2 h, until the solution had become colourless. Water (25 cm3) was added resulting in the formation of a white precipitate. After boiling the mixture for 10 min, the precipitate was filtered and re-dissolved in chloroform, dried over magnesium sulphate, filtered and all volatiles removed under reduced pressure to afford a white solid. Recrystallisation from toluene afforded the title compound as white crystals. Yield: 1.01 g, 39%. Found C: 72.7%, H: 8.1%, N: 8.6% and C20H26N2O2 requires C: 73.6%, H: 8.0%, N: 8.6%. νmax = 3293 (NH stretching) cm–1. m/z (EI) = 327 [M + H]+. δ1H (CDCl3): 7.19 (m, 2H, C8H), 6.96 (m, 2H, C9H), 6.80 (m, 2H, C6H), 6.74 (m, 2H, C7H), 4.01 (d, 2H, 2JHH = 13.3 Hz, C4a/bH, C4'a/bH), 3.94 (d, 2H, 2JHH = 13.3 Hz, C4a/bH, C4'a/bH), 2.43 (m, 2H, C3H, C3′H), 2.12 (m, 4H, C2H, C2'H), 1.41 (m, 2H, C1H, C1′H), 1.23 (m, 4H, C1/2H, C1/2′H). δ13C (CDCl3): 157.96 (s, 2C, C10, C10'), 128.82 (s, 2C, C6, C6'), 128.28 (s, 2C, C7, C7'), 122.89 (s, 2C, C5, C5'), 119.21 (s, 2C, C9, C9'), 116.43 (s, 2C, C8, C8'), 59.72 (s, 2C, C3, C3'), 49.69 (s, 2C, C4, C4'), 30.44 (s, 2C, C2, C2'), 24.14 (s, 2C, C1, C1').
4.3 Synthesis of (R,R)-tBu4-salcyan 2
(R,R)-tBu4-salcyen (1.04 g, 1.9 mmol) was dissolved in ethanol (75 cm3) and heated at gentle reflux for 20 min. To this mixture, NaBH4 (0.45 g, 11 mmol) was added in small portions and gentle reflux was maintained until the solution became colourless. At this point water (75 cm3) was added to reveal a white precipitate, which was collected by reduced pressure filtration and recrystallised from pentane, to afford the title compound as white crystals. Yield: 1.76 g, 60%. Found C: 78.6%, H: 10.5%, N: 5.1% and C36H58N2O2 requires C: 78.5%, H: 10.6%, N: 5.1%. νmax = 3293 (NH stretching), 1604 (NH deformation) cm–1. m/z (EI) = 550 [M]+, 332 [M – (C6H2(tBu)2CH2O)]. δ1H (CDCl3): 10.6 (broad s, 2H, NH/OH), 7.20 (d, 2H, JHH = 2.4 Hz, C8H, C8′H), 6.86 (d, 2H, JHH = 2.4 Hz, C6H, C6′H), 4.01 (d, 2H, 2JHH = 13.3 Hz, C4a/bH, C4′a/bH), 3.92 (d, 2H, 2JHH = 13.3 Hz, C4a/bH, C4′a/bH), 2.46 (m, 2H, C3H, C3′H), 2.17 (m, 4H, C2H, C2′H), 1.71 (s, 4H, C1H, C1′H), 1.37 (s, 18H, C14H, C14′H), 1.28 (s, 18H, C12H, C12′H). δ13C (CDCl3): 154.37 (s, 2C, C10, C10′), 140.67 (s, 2C, C7, C7′), 136.03 (s, 2C, C9, C9′), 123.13 (s, 2C, C6, C6′), 123.02 (s, 2C, C8, C8′), 122.40 (s, 2C, C5, C5′), 59.91 (s, 2C, C3, C3′), 50.85 (s, 2C, C4, C4′), 34.86 (s, 2C, C11, C11′), 34.14 (s, 2C, C13, C13′), 31.67 (s, 2C, C12, C12′), 30.73 (s, 2C, C2, C2′), 29.63 (s, 2C, C14, C14′), 24.13 (s, 2C, C1, C1′).
4.4 Synthesis of [(R,R)-salcyan]Al(OH) 3
(R,R)-salcyan 1 (1.0 g, 2.02 mmol) was dissolved in toluene (20 cm3) and stirred whilst trimethylaluminium (2.0 M solution in hexanes, 1.38 cm3, 2.02 mmol) was added slowly. The reaction mixture was stirred at 25 °C for 5 h, after which time the mixture was exposed to air and the solvent removed. The crude material was recrystallised from dichloromethane: pentane (1:1 v/v) to afford the title compound as an off-white crystalline solid. Yield: 0.28 g, 40%. M.p.; 267-268 °C. Found C: 64.5%, H: 7.0%, N: 6.9% and AlC20H25N2O3 requires C: 65.2%, H: 6.8%, N: 7.6%. m/z (ES; 1:1 v/v H2O:MeCN) = 351 [SalcyanAl]+.
4.5 Synthesis of [(R,R)-tBu4-salcyan]Al(OH) 4
(R,R)-tBu4-salcyan 2 (0.22 g, 0.40 mmol) was dissolved in pentane (5 cm3) and trimethylaluminium (2.0 M solution in hexanes, 0.20 cm3, 0.40 mmol) added with stirring. Stirring was continued for 5 h, after which time the mixture was exposed to air and the solvent removed. The crude (0.31 g) was recrystallised from pentane to afford the title compound as white crystals. Yield: 0.23 g, 50%. M.p.; 282–284 °C. Found C: 72.9%, H: 9.9%, N: 4.5% and AlC36H57N2O3 requires C: 72.9%, H: 9.7%, N: 4.7%. m/z (ES; methanol) = 1212 [M–OMe]2+, 615 [M–OH]Na+, 629 [M – OMe]Na. δ1H (CDCl3): 7.31 (d, 2H, C6H/C6′H), 7.10 (d, 2H, C8H/C8′H), 6.72 (dd, 4H, C6H/C6′H, C8H C8′H), 4.74 (m, 2H, NH, N′H), 4.64 (d, 2H, C4a/bH/C4a/b′H), 3.78 (d, 2H, C4a/bH/C4a/b′H), 3.65 (d, 2H, C4a/bH/C4a/b′H), 3.58 (d, 2H,C4a/bH/ C4a/b′H), 2.65 (d, 2H, C3H/C3′H), 2.37 (d, 2H, C2H/C2′H), 2.18 (d, 2H, C3H/C3′H), 1.77 (d, 2H, C1H/C1′H), 1.62 (d, 2H, C1H/C1′H), 1.58 (s, 9H, C14H/C14′H, tBu), 1.27 (s, 9H, C12H/C12′H, tBu), 1.24 (s, 9H, C12H/C12′H, tBu), 1.08 (m, 2H, C2H/C2′H), 0.91 (s, 9H, C14H/C14′H, tBu). δ13C (CDCl3): 160.43, 158.02 (s, 2C, C10/C10′), 138.22, 138.17 (s, 2C, C/ C7′), 137.31, 136.43 (s, 2C, C9/C9′), 124.78, 124.74 (s, 2C, C6/C6′), 123.38, 123.35 (s, 2C, C8/C8′), 123.08, 121.85 (s, 2C, C3/C3′), 59.01, 56.68 (s, 2C, C3/C3′), 50.99, 49.17(s, 2C, C4/C4′), 35.38,34.76, 33.93, 33.80 (s, 2C, C11/C11′) or (s, 2C, C13/C13′), 31.78, 31.75, 31.26, 29.81 (s, 2C, C12/C12′) or (s, 2C, C1/ C14′), 29.84, 29.21 (s, 2C, C/C2′), 24.71, 24.53 (s, 2C, C1/C1′).
4.6 Phospho-aldol catalysis with salcyen and salcyan complexes
H-Phophonate (1 mmol) and benzaldehyde (or para-substituted benzaldehyde, 1 mmol) were added to a stirred solution of the catalyst precursor (x mol%) in dry distilled THF (4 cm3) under an aerobic atmosphere in a vessel maintained at the temperature of interest. The mixture was subsequently, stirred at that temperature for a period of 3–6 h. After the required reaction time, all volatile materials were removed under reduced pressure and the residue dissolved in CDCl3 and analysed first by 31P{1H}–NMR spectroscopy to ascertain the extent of reaction during the period studies and then secondly, re-analysed in the presence of quinine (4 mmol) from which enantioselectivities were readily calculated as described previously [11].
4.7 Crystallography
The asymmetric unit of complex 4 contains one molecule of the complex and four half-occupied cyclohexane environments, all lying on general positions. Three of the four solvent sites are disordered, containing two distinct partial molecules with equal occupancies of 0.25. All C–C bond lengths and 1,3-CC distances within the six 0.25-occupied solvent molecules were restrained to 1.51(2)\%A and 2.47(2)\%A, respectively. All non-H atoms with occupancy ≥ 0.5 were refined anisotropically. All H atoms were placed in calculated positions and refined using a riding model. Attempts to allow the positions of the hydroxide atoms H3 and H4 to freely refine with a restrained O–H distance were unsuccessful, and these were therefore placed in calculated positions corresponding to idealised trigonal geometries for the corresponding O atoms. Data for compound 2 were collected at 150(2) K on a colourless prismatic crystal mounted under oil on a Nonius KappaCCD with Mo Kα radiation (λ = 0.71073 Å). Data reduction was performed using DENZO and data were corrected for Lorentzian and polarisation effects but not absorption. The structure was solved by direct methods with SHELXS-97 and refined with full-matrix least squares on F2 with SHELXL-97. All non-hydrogen atoms were refined anisotropically. Carbon positions within each phenyl ring were restrained to be coplanar using the FLAT command of SHELXL-97. Hydrogen atoms were included at calculated positions using a riding refinement. Further details of the data collection on both 2 and 4 along with structure refinement details are given in Table 9.
Crystal data and structure refinements for compounds 2 and 4
| Identification code | Compound 4 |
| Formula | C72H112Al2N4O62 C6H12 |
| Formula weight | 1353.94 |
| Size | 0.40 × 0.33 × 0.33 mm |
| Crystal morphology | Colourless prism |
| Wavelength | 0.71073 Å [Mo Kα] |
| Crystal system | Monoclinic |
| Space group | P21 (No. 4) |
| Unit cell dimensions | a = 12.4844(7) Å, α = 90° |
| b = 22.4069(14) Å, β = 107.390(3)° | |
| c = 17.5985(9) Å, γ = 90° | |
| Volume | 4697.9(5) Å3 |
| Z | 2 |
| Absorption coefficient | 0.076 mm–1 |
| F(000) | 1216 |
| Data collection range | 4.9 ≤ θ ≤ 21.5° |
| Index ranges | –15 ≤ h ≤15, –17 ≤ k ≤ 17, –25 ≤ l ≤ 25 |
| Independent reflections | 9327 [R(int) = 0.061] |
| Observed reflections | 7667 [I >2σ(I)] |
| Absorption correction | multi-scan |
| Max. and min. transmission | 0.9887 and 0.9715 |
| Refinement method | Full |
| Data / restraints / parameters | 9327/73/956 |
| Goodness of fit | 1.075 |
| Final R indices [I > 2 σ(I)] | R1 = 0.070, wR2 = 0.191 |
| Largest diff. peak and hole | 0.81 and –0.24 e Å-3 |
| Absolute structure parameter | 0.0(3) |
| Identification code | Compound 2 |
| Formula | C36H58N2O2 |
| Formula weight | 550.84 |
| Size | 0.46 × 0.19 × 0.18 mm |
| Crystal morphology | Colourless prism |
| Temperature | 150(2) K |
| Wavelength | 0.71073 Å [Mo Kα] |
| Crystal system | Orthorhombic |
| Space group | P212121 |
| Unit cell dimensions | a = 12.27910(10) Å, α = 90° |
| b = 13.9327(2) Å, β = 90° | |
| c = 20.5380(3) Å, γ = 90° | |
| Volume | 3513.66(8) Å3 |
| Z | 4 |
| Density (calculated) | 1.041 Mg m–3 |
| Absorption coefficient | 0.063 mm–1 |
| F(000) | 1216 |
| Data collection range | 2.21 ≤ θ ≤ 26° |
| Index ranges | –15 ≤ h ≤15, –17 ≤ k ≤ 17, –25 ≤ l ≤ 25 |
| Reflections collected | 52339 |
| Independent reflections | 6868 [R(int) = 0.0628] |
| Observed reflections | 6309 [I > 2 σ(I)] |
| Absorption correction | multi-scan |
| Max. and min. transmission | 0.9887 and 0.9715 |
| Refinement method | full |
| Data / restraints / parameters | 6868/0/390 |
| Goodness of fit | 1.075 |
| Final R indices [I > 2 σ(I)] | R1 = 0.0584, wR2 = 0.1565 |
| R indices (all data) | R1 = 0.0644, wR2 = 0.164 |
| Largest diff. peak and hole | 0.468 and –0.256 e Å–3 |
| Absolute structure parameter | 0.0(15) |
Acknowledgements
We thank the EPSRC for a studentship to T.D.N. (GR/R39689), the China Scholar Council for a Fellowship to M.J and Messrs. Ivan Litvinenko-Morel and Kevin Mercier for valuable synthetic help.