

1 Introduction
N-Hydroxylamines are a very interesting class of compounds in organic chemistry, because of their interest as intermediates in the preparation of highly functionalized amino derivatives. They may be prepared by reaction of nitrones with nucleophiles [1–15]. So, we have previously shown that N-hydroxylamines, obtained by reaction of nitrones with alkyl 3-lithiopropiolates, could be transformed into the corresponding γ-amino α,β-saturated esters [12], γ-(N-benzyl)amino α,β-ethylenic esters and/or α,β-ethylenic γ-lactames [3], γ-(N-t-butoxycarbonyl) amino α,β-ethylenic esters [6] and γ-(N-t-butoxycarbonyl)amino α,β-dihydroxy esters [15]. The study of the reactivity of nitrones with indolic rings was also carried out [16,17], leading to the development of a synthetic approach of indolic N-hydroxylamines (Fig. 1 ). In this paper, we describe the preparation, by this method, of (R, 4R)-3-[(N-benzyl-N-hydroxy-amino)-(1H-indol-3-yl)-methyl]-oxazolidin-2-one 2a and its (S, 4R) diastereomer 2b from α-chiral nitrone 1a and indole [18] (Fig. 1). We also disclose the crystal structure analysis of compound 2a (Fig. 2 ).
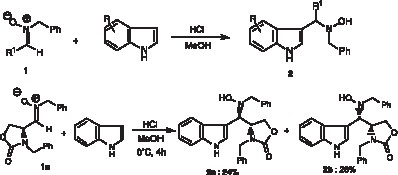
Preparation scheme of the title compound.
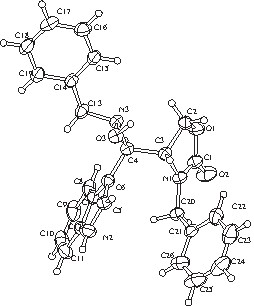
Perspective view of the title compound performed using ORTEP II.
2 Preparation
The experimental procedure is as follows.
To a stirred methanolic solution of anhydrous hydrochloric acid (obtained by addition of 0.461 ml (0.506 g, 6.45 mmol) of acetyl chloride in 28 ml of distilled methanol), at 0 °C and under argon, was added a solution of 1.0 g (3.22 mmol) of nitrone 1a and 0.377 g (3.22 mmol) of indole in 7 ml of distilled methanol. The resulting mixture was stirred at 0 °C for 4 h and then diluted by addition of dichloromethane. A saturated aqueous solution of sodium hydrogencarbonate was then added. After decantation, the aqueous layer was extracted three times with acetyl acetate. The combined organic layers were washed with brine and dried over anhydrous magnesium sulfate. After filtration and evaporation of solvents under vacuum, the crude product was purified by silica gel chromatography using a mixture of ether:pentane (1:7) as eluent to afford 0.331 g (0.78 mmol) of pure N-hydroxylamine 2a and 0.379 g (0.89 mmol) of its diastereomer 2b as white solids. The yields are respectively 24% and 28%.
Fig. 1 schematizes the experimental process for the preparation of indolic N-hydroxylamines 2a and 2b.
Experimental data for 2a. mp : 88 °C. [α]D20 = –5.1 (c = 1.1; CHCl3). 1H NMR (300 MHz, CDCl3) δ 3.72 (ABq, JAB = 13.0 Hz, δA – δB = 87.1 Hz, 2H); 3.80–3.98 (broad s, 1H); 4.15–4.30 (m, 3H); 4.51 (broad s, 1H); 4.67 (s, 1H); 4.87 (d, J = 14.9 Hz, 1H); 6.86–7.00 (m, 2H); 7.04–7.14 (m, 1H); 7.16–7.37 (m, 11H); 7.40 (d, J = 8.1 Hz, 1H); 8.54 (broad s, 1H). 13C NMR (75.5 MHz, CDCl3) δ 47.1 (CH2); 55.8 (CHN); 62.3 (CH2); 63.2 (CHN); 65.4 (CH2); 108.6 (C); 111.5 (CH); 118.4 (CH); 120.1 (CH); 122.5 (CH); 124.3 (CH); 127.5 (CH); 127.6 (C); 127.7 (2 CH); 128.4 (CH); 128.7 (CH); 129.4 (CH); 135.7 (C); 136.1 (C); 137.4 (C); 159.5 (CO). IR (film): 3410 and 3300 (νNH and νOH), 1730 cm–1 (νCO oxazolidinone). Mass spectrum (DCI, NH3 + isobutane) m/z 428 (MH+), 305. Anal. calcd for C26H25N3O3: C, 73.05; H, 5.89; N, 9.83; found: C, 72.91; H, 6.17; N, 9.64. Experimental data for 2b. mp: 88 °C. [α]D20 = –23.7 (c = 0.75; CHCl3). 1H NMR (300 MHz, CDCl3) δ 3.64 (ABq, JAB = 13.0 Hz, δA – δB = 16.1 Hz, 2H); 3.85–4.03 (m, 2H); 4.11 (broad s, 1H); 4.37–4.50 (m, 2H); 4.90 (ABq, JAB = 15.0 Hz, δA – δB = 93.8 Hz, 2H); 6.85–7.40 (m, 14H); 7.54 (d, J = 7.8 Hz, 1H); 8.60 (broad s, 1H). 13C NMR (75.5 MHz, CDCl3) δ 47.1 (CH2); 54.7 (CHN); 62.3 (CH2); 65.4 (CH2); 66.3 (CHN); 107.3 (C); 111.5 (CH); 119.0 (CH); 120.3 (CH); 122.5 (CH); 124.5 (CH); 127.5 (CH); 127.8 (2 CH); 128.0 (C); 128.4 (CH); 129.0 (CH); 129.4 (CH); 135.7 (C); 136.8 (C); 137.3 (C); 159.3 (CO). IR (film): 3420 and 3340 (νNH and νOH), 1730 cm–1 (νCO oxazolidinone). Mass spectrum (DCI, NH3 + isobutane) m/z 428 (MH+), 305.
3 Structural determination
A crystal fragment with a size of 0.18 × 0.12 × 0.10 mm3 was used for the collection of diffraction data run with an Enraf-Nonius CAD4 diffractometer, operating with the copper radiation (1.5418 Å), monochromatized by a graphite plate. Measurements were performed within a range of 3 to 72° (θ). In this explored area (±h, k, l) h varies from –10 to 10, k from 0 to 15 and l from 0 to 25. A total of 4560 independent reflections (Rint = 0.048) were extracted among the 4599 scanned reflections. All reflections were measured with an ω/2θ scan within an angular range of 1.20° for a maximum time of 80 s. Two intensity reference reflections measured every two hours showed a decay of 6.74% during measurements run at room temperature. Unit-cell dimensions were determined and refined from a set of 25 reflections selected between 21.6 and 27.10°(θ). Some additional data are: Dx = 1.259, F(000) = 904, V = 2256(1) Å3, μ = 0.672 cm–1.
The observed extinction rules: 0k0 (k = 2 n+1) and h0l (l = 2 n+1) lead unambiguously to the P21/c space group. No absorption correction was applied. The crystal structure was solved using a direct method [19]. All non-hydrogen atoms were refined anisotropically, while hydrogen atoms located by geometry were not. The weighting scheme used corresponds to w = [σ2(F0) + p2/4|F0|2]–1 with p = 0.06. For 3584 reflections corresponding to I/σ(I) > 2, the final R value is 0.052 (Rw = 0.083). The residual electronic densities in the final Fourier difference map spread between –0.18 and 0.19 e Å–3. The final atomic coordinates and the equivalent thermal factors are gathered in Table 1. All calculations as well as the ORTEP view [20] were made using the TeXsan cristallographic system [21].
Atomic coordinates and thermal parameters. Estimated standard deviations are given in parentheses
| Atoms | x(σ) | y(σ) | z(σ) | Beq. |
| O(1) | 1.1940(1) | 0.13858(9) | 0.77428(6) | 0.0636(3) |
| O(2) | 1.1957(2) | 0.2977(1) | 0.72608(7) | 0.0820(4) |
| O(3) | 0.6986(1) | –0.01914(8) | 0.83677(5) | 0.0537(3) |
| N(1) | 0.9727(2) | 0.2017(1) | 0.73478(6) | 0.0495(3) |
| N(2) | 0.4992(2) | 0.2832(1) | 0.79437(8) | 0.0674(4) |
| N(3) | 0.8307(1) | 0.03136(9) | 0.86924(6) | 0.0459(3 |
| C(1) | 1.1241(2) | 0.2200(1) | 0.74288(8) | 0.0575(4) |
| C(2) | 1.0840(2) | 0.0531(1) | 0.77946(8) | 0.0560(4) |
| C(3) | 0.9281(2) | 0.1063(1) | 0.77047(7) | 0.0458(3) |
| C(4) | 0.8559(2) | 0.1333(1) | 0.83546(7) | 0.0441(3) |
| C(5) | 0.5835(2) | 0.1911(1) | 0.79550(8) | 0.0559(4) |
| C(6) | 0.7213(2) | 0.2072(1) | 0.82797(7) | 0.0470(4) |
| C(7) | 0.7216(2) | 0.3167(1) | 0.84789(7) | 0.0496(4) |
| C(8) | 0.8296(2) | 0.3824(1) | 0.88121(8) | 0.0616(5) |
| C(9) | 0.7920(3) | 0.4869(2) | 0.8910(1) | 0.0827(6) |
| C(10) | 0.6532(3) | 0.5293(2) | 0.8686(1) | 0.1000(7) |
| C(11) | 0.5456(3) | 0.4681(2) | 0.8363(1) | 0.0860(6) |
| C(12) | 0.5819(2) | 0.3615(1) | 0.82592(8) | 0.0616(4) |
| C(13) | 0.7872(2) | 0.0493(1) | 0.93606(7) | 0.0559(4) |
| C(14) | 0.7817(2) | –0.0515(1) | 0.97540(7) | 0.0504(4) |
| C(15) | 0.8638(2) | –0.1418(1) | 0.96092(8) | 0.0632(5) |
| C(16) | 0.8577(3) | –0.2313(1) | 0.99942(9) | 0.0740(6) |
| C(17) | 0.7702(3) | –0.2323(2) | 1.05295(9) | 0.0737(6) |
| C(18) | 0.6882(2) | –0.1434(2) | 1.06819(8) | 0.0743(6) |
| C(19) | 0.6933(2) | –0.0536(1) | 1.02971(8) | 0.0630(5) |
| C(20) | 0.8732(2) | 0.2754(1) | 0.69864(7) | 0.0546(4) |
| C(21) | 0.7832(2) | 0.2280(1) | 0.64241(7) | 0.0562(4) |
| C(22) | 0.8292(2) | 0.1377(2) | 0.61079(9) | 0.0776(6) |
| C(23) | 0.7483(3) | 0.1016(2) | 0.5557(1) | 0.0988(8) |
4 Supplementary material available
Hydrogen coordinates, anisotropic thermal parameters, tables of interatomic distances and bond angles involving hydrogen atoms are available. Supplementary material data have been deposited at the Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB2 1EZ, UK, as supplementary publication No. 216151 and are available on request from the CCDC.
Acknowledgements
This work has been generously supported by the French ‘Association pour la recherche contre le cancer’ (ARC).