

1 Introduction
Trimeric perfluoro-ortho-phenylene mercury (1) [1] is a simple trifunctional Lewis acid that has been used for the complexation of both anionic and neutral Lewis basic substrates [2–7]. In a series of studies, we also demonstrated that this trinuclear mercury derivative readily complexes with aromatic substrates including benzene, naphthalene, biphenyl, pyrene and triphenylene [8–11]. The resulting adducts consists of supramolecular stacks in which the arene interacts with the mercury centers of 1 through secondary mercury-π interactions. It occurred to us that similar structures might result from the interaction of 1 with more π−basic molecules. As a result, we have investigated its interaction with tetrathiafulvalene (TTF). As part of this work and in order to shed further light on the chemical affinity of 1, we have also studied its interaction with the typical π-acidic 7,7,8,8-tetracyanoquinodimethane (TCNQ). These efforts were further motivated by a series of recent investigations in which it has been demonstrated π−basic trinuclear gold(I) derivatives interact with various π-acidic molecules including C6F6, TCNQ [12] and nitrofluorenones [13].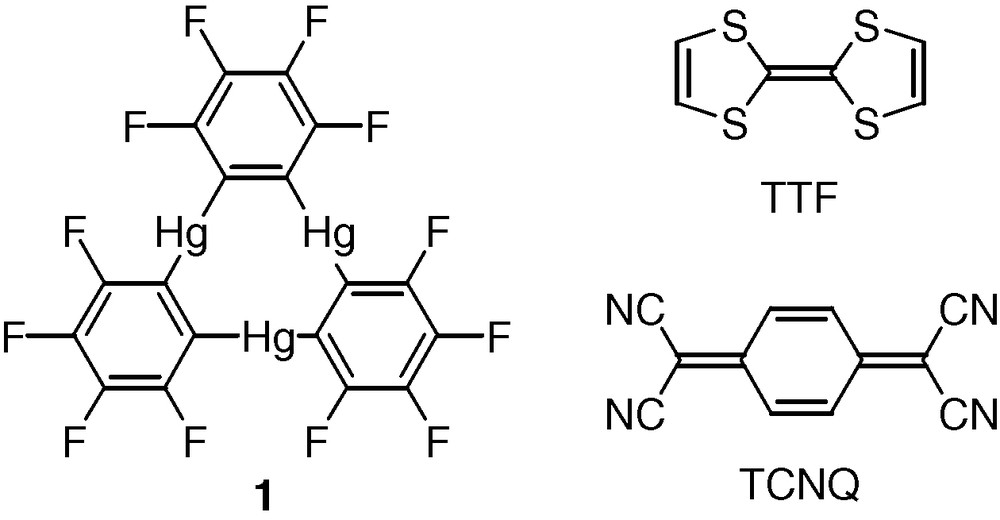
2 Results and discussion
When 1 and TTF are combined in a 1:1 mixture of CH2Cl2 and CS2, slow evaporation of the solvent leads to the formation of orange needles of the 2:1 adduct [(1)2·TTF] (2). Interestingly, a similar experiment carried out with TCNQ affords light yellow needles of an adduct of identical stoichiometry, namely [(1)2·TCNQ] (3). In both cases, the color of these complexes corresponds to that of the organic molecule which rules out the presence of intense charge transfer bands. The 199Hg, 19F, and 1H NMR spectra of these compounds in CD2Cl2 as well as their UV absorption spectra correspond to those of the free molecular components which indicates complete dissociation of the adducts upon dissolution.
Compound 2 crystallizes in the triclinic space group with one centrosymmetrical [(1)2·TTF] adduct per unit cell (Table 1, Fig. 1). Examination of the structure indicates that each molecule of TTF is sandwiched by two molecules of 1 and engages in multiple Hg···S secondary interactions. Taking into account the range of values that have been suggested for the van der Waals radii of both mercury (rvdw = 1.73–2.00 Å) [14,15] and sulfur (rvdw = 1.8–2.03 Å) [16–19], the length of these interactions appears to be either close to or shorter than the sum of the van der Waals radii of the two elements and mercury. As shown in Fig. 1, the S(1) and S(2) atoms coordinate to the Hg(3) mercury center in a bidentate fashion (Hg(3)–S(1) 3.467(5) Å, Hg(3)-S(2) 3.533(5) Å). The sulfur atom S(1) forms an additional interaction with the mercury center Hg(2) (Hg(2)–S(1A) 3.529(5) Å). These secondary interactions are comparable to those found in [1·SCN]– [2] and [1·μ6-SMe2]n [20]. They are also respectively longer and shorter than the primary Hg–S bond (2.40 Å) and the secondary Hg···S interactions (3.89 Å) observed in the structure of [tht·HgCl2]. The structure of [tht·HgCl2] features short intramolecular primary Hg–S bonds and long intermolecular secondary Hg–S interactions [21]. It is interesting to point to the structural resemblance that exists between 2 and a series of gold-containing supramolecules reported by Balch [13] as well as Burini and Fackler [22]. As mentioned earlier, these supramolecules consist of stacks in which trinuclear gold(I) complexes alternate with organic derivatives such as fluorenones, hexafluorobenzene, and TCNQ [12]. At the difference of 2, the trinuclear gold(I) complexes are electron-rich while the organic substrates are electron-poor. Compound 2, which contains electron-poor mercury centers and electron-rich TTF molecules, can therefore be regarded as the charge-reverse analogue of the gold assemblies.
Crystal data, data collection, and structure refinement for 2 and 3–(CS2)3
| Crystal data | 2 | 3–(CS2)3 |
| Formula | C42H4F24Hg6S4 | C51H4F24Hg6N4S6 |
| Mr | 2296.23 | 2524.48 |
| Crystal size (mm3) | 0.29 × 0.055 × 0.055 | 0.31 × 0.15 × 0.080 |
| Crystal system | Triclinic | Triclinic |
| Space group | P | P |
| a (Å) | 8.9347(18) | 8.9533(18) |
| b (Å) | 10.017(2) | 12.038(2) |
| c (Å) | 13.669(3) | 14.641(3) |
| α (°) | 87.57(3) | 96.47(3) |
| β (°) | 75.12(3) | 96.35(3) |
| γ (°) | 78.75(3) | 109.82(3) |
| V (Å3) | 1159.5(4) | 1456.3(5) |
| Z | 1 | 1 |
| ρcalc (g cm–3) | 3.288 | 2.878 |
| μ(Mo Kα) (mm–1) | 20.098 | 16.088 |
| F(000) (e) | 1016 | 1130 |
| Data collection | ||
| T (K) | 293(2) | 293(2) |
| Scan mode | ω | ω |
| hkl range | –10→9, –11→11, –16→16 | –10→10, –14→14, –17→16 |
| Measured refl. | 11 377 | 13 915 |
| Unique refl., [Rint] | 4068 [0.0486] | 5088 [0.0286] |
| Refl. used for refinement | 4068 | 5088 |
| Absorption correction | SADABS | SADABS |
| Tmin/Tmax | 0.122867 | 0.315385 |
| Refinement | ||
| Refined parameters | 343 | 412 |
| R1, wR2 [I > 2 σ(I)] | 0.0443, 0.1168 | 0.0460, 0.1091 |
| ρfin (max/min) (e Å–3) | 2.319, –1.982 | 4.332, –1.558 |
| a R1 = (Fo – Fc)/Fo.b wR2 = { [w(Fo2 – Fc2)2]/[w(Fo2)2]}1/2; w = 1/[σ2(Fo2) + (ap)2 + bp]; p = (Fo2 + 2 Fc2)/3; a = 0.0851 (2), 0.0808 (3); b = 0 (2), 15.0890 (3). |
Crystal structure of 2. Mercury (orange), sulfur (yellow), fluorine (green), and carbon (black). Intramolecular distances (Å): Hg(2)–S(1) 3.529(5), Hg(3)–S(2) 3.533(5), Hg(3)–S(1A) 3.467(5). Intermolecular bond distances (Å) and angles (º): Hg(1)–C(8) 2.111(14), Hg(1)–C(1) 2.122(14), Hg(2)–C(7) 2.051(13), Hg(2)–C(14) 2.086(15), Hg(3)–C(2) 2.041(16), Hg(3)–C(13) 2.069(15), C(20)–C(20A) 1.37(2), S(1)–C(20) 1.779(13), S(2)–C(20) 1.729(13), C(8)–Hg(1)–C(1) 174.8(6), C(7)–Hg(2)–C(14) 176.4(7), C(2)–Hg(3)–C(13) 174.4(6), S(2)–C(20)–S(1) 114.4(7).
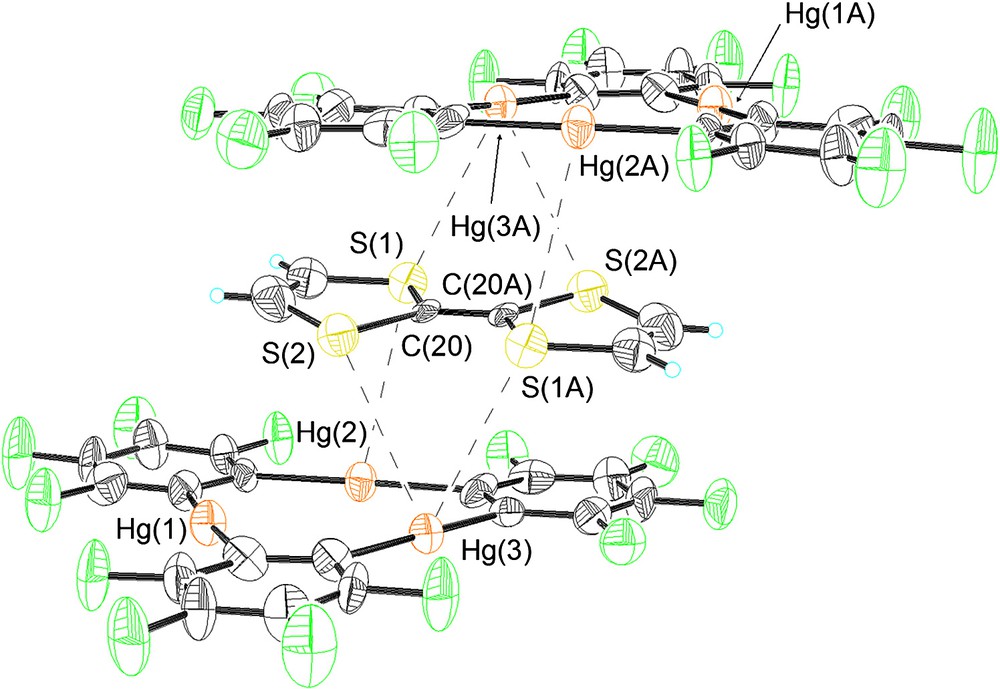
As in the case of 2, compound 3 crystallizes in the triclinic P space group with one centrosymmetrical [(1)2·TCNQ] adduct per unit cell that also contains three molecules of CS2 (Table 1, Figs. 2–4). Examination of the atomic connectivity reveals the simultaneous coordination of one of the nitrile nitrogen atoms to the three mercury centers of 1 (Fig. 1). The resulting Hg–N distances range from 3.102(11) to 3.134(11) Å and are well within the sum of the van der Waals radii for mercury (rvdw = 1.73–2.00 Å) [14,15] and nitrogen (rvdw = 1.60 Å). As a result of these interactions, the nitrogen atom (N(1)) is essentially equidistant from the three Lewis acidic sites and sits at 2.32 Å from the plane defined by the three mercury atoms. The linear nitrile functionality is almost perpendicular to the plane of the trinuclear complex with which it forms an angle of 88.7°. The metrical and angular parameters observed in 3 resemble those encountered in the acetonitrile adduct [1·(μ3-acetonitrile)2] [23,24] (avg. Hg–N = 2.96 Å). Adducts involving acetonitrile and mercuracarborands have also been isolated [25]. The trans nitrile group of the TCNQ molecules coordinates to another molecule of 1 thus completing the [(1)2·TCNQ] unit. The remaining two nitrile groups of the TCNQ molecule do not engage in any donor interactions. Three molecules of CS2 are trapped between neighboring [(1)2·TCNQ] units and interact with the mercury centers of the juxtaposed molecules of 1 (Fig. 3). Two of these molecules are symmetrically equivalent and are terminally ligated to the mercury center Hg(2) (Hg(2)–S(2) 3.532(6) Å). The third molecule of CS2 is centrosymmetrical and interacts via each of its two sulfur atoms with four mercury centers provided by the two molecules of 1 (Hg(2)–S(3) 3.640(7) Å, Hg(3)–S(3) 3.485(6) Å). These Hg···S distances are comparable to those observed in the structure of 2 and are once again comparable or shorter than the sum of the van der Waals radii of the two elements. As a result of these interaction, the solid-state structure of 3–(CS2)3 consists of extended one-dimensional chain, as depicted in Fig. 4.
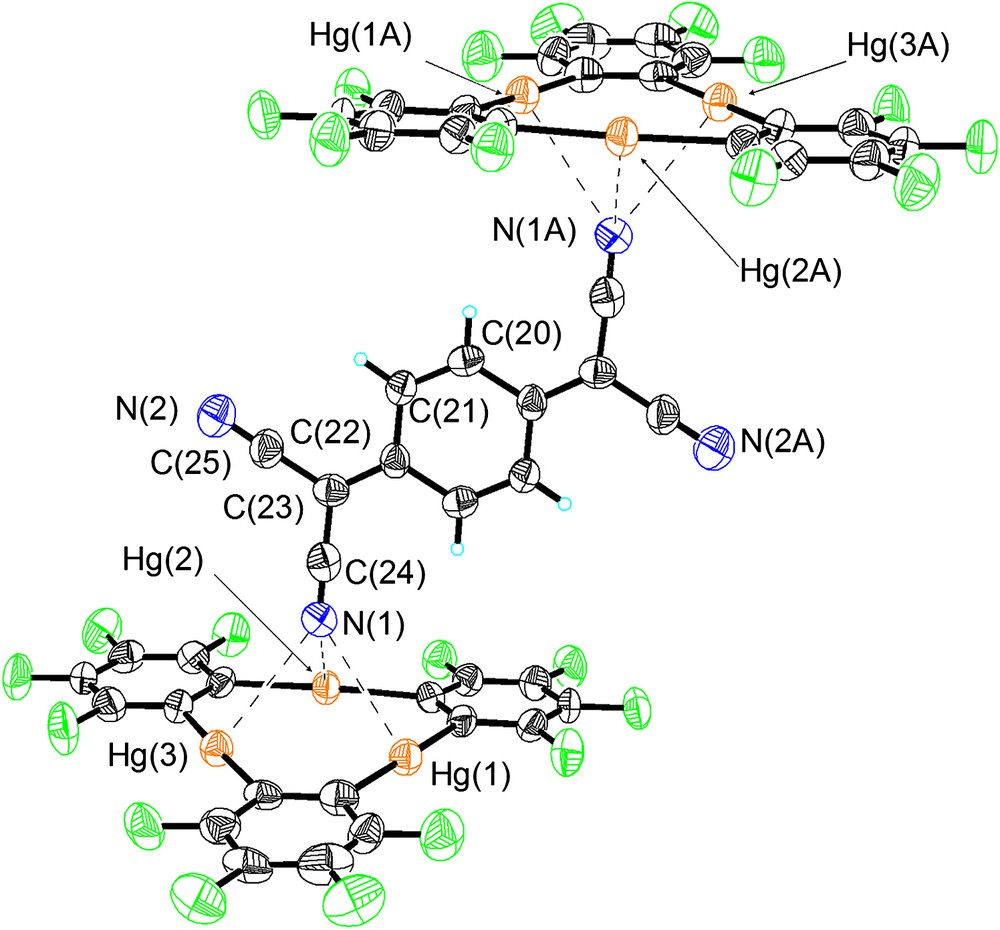
Crystal structure of 3 illustrating the Hg–N interactions. Mercury (orange), fluorine (green), nitrogen (blue), carbon (black). Intramolecular distances (Å): Hg(1)–N(1) 3.102(11), Hg(2)–N(1) 3.128(12), Hg(3)–N(1) 3.134(11). Intermolecular bond distances (Å) and angles (°): Hg(1)–C(1) 2.089(14), Hg(1)–C(8) 2.099(12), Hg(2)–C(14) 2.104(12), Hg(2)–C(7) 2.109(12), Hg(3)–C(13) 2.079(14), Hg(3)–C(2) 2.079(13), N(1)–C(24) 1.157(17), N(2)–C(25) 1.130(18), C(20)–C(21) 1.335(18), C(21)–C(22) 1.438(17), C(22)–C(23) 1.366(17), C(23)–C(24) 1.424(18), C(23)–C(25) 1.449(18), C(1)–Hg(1)–C(8) 175.3(5), C(14)–Hg(2)-C(7) 176.0(5), C(13)–Hg(3)–C(2) 176.2(5), C(24)–C(23)–C(25) 116.1(11), N(1)–C(24)–C(23) 178.6(15), N(2)–C(25)–C(23) 177.8(17).
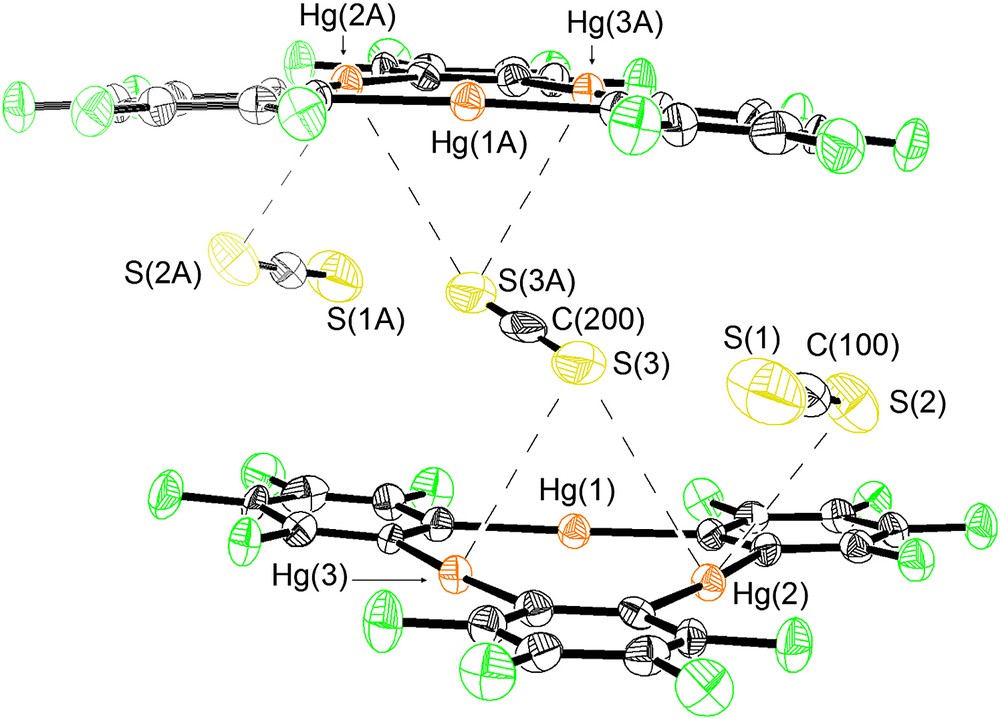
Crystal structure of 3 illustrating the Hg–S interactions. Mercury (orange), fluorine (green), sulfur (yellow), carbon (gray). Intramolecular distances (Å): Hg(2)–S(2) 3.532(6), Hg(2)–S(3) 3.640(7), Hg(3)–S(3) 3.485(6). Intermolecular bond distances (Å) and angles (°): S(1)–C(100) 1.57(3), S(2)–C(100) 1.51(2), S(3)–C(200) 1.553(11), S(2)–C(100)–S(1) 174.6(18), S(3)–C(200)–S(3A) 180.0(16).
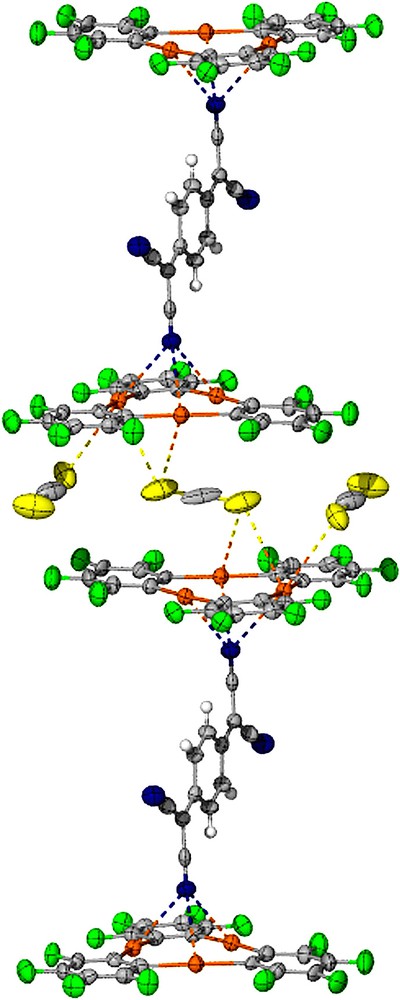
Extended structure of 3. Mercury (orange), fluorine (green), nitrogen (blue), sulfur (yellow), carbon (gray).
3 Conclusions
The results reported herein further document the acceptor ability of 1. While known π–donors such as TTF coordinate to 1 in a stacking fashion, known π– acceptors such as TCNQ interact with 1 through the electron rich terminus of the nitrile groups. These results clearly indicate that compound 1 is a Lewis acid. It is also important to note that the spectroscopic and structural results presented herein do not support oxidation of the TTF molecules. As a result, compound 2 cannot be described as a charge transfer salt. Rather, its cohesion apparently results from the presence of secondary donor interaction occurring between the sulfur atoms of TTF and the mercury centers of 1.
4 Experimental
4.1 Materials and methods
Due to the toxicity of the mercury compounds discussed in these studies, extra care was taken at all times to avoid contact with solid, solution, and airborne particulate mercury compounds. The studies herein were carried out in well-aerated fume hood. The infrared spectra were recorded as KBr pellets on a Mattson Genesis Series FTIR. Atlantic Microlab, Inc., Norcross, GA, performed the elemental analyses. TTF was purchased from TCI America and used as provided. Other commercially available starting materials and solvents were purchased from Aldrich Chemical and were used as provided. Compound 1 was prepared according to the published procedure outlined by Sartori and Golloch [1].
4.2 Synthesis of 12·TTF (2)
Compound 1 (100 mg, 9.6 μmol) was dissolved in a 1:1 mixture of CS2 and CH2Cl2 (30 ml). In a separate vial TTF (10 mg, 5.1 μmol) was dissolved in a 1:1 mixture of CS2 and CH2Cl2 (3 ml). The two solutions were mixed thoroughly. Partial evaporation of the solvent resulted in the crystallization of 2, which was isolated in a 70% yield (153 mg, 6.7 μmol). mp decomposition 285 °C. Anal. calc. (found) for C42H4F24Hg6S4: C, 21.96 (22.16); H, 0.18 (0.16).
4.3 Synthesis of 12·TCNQ·3CS2 (3–(CS2)3)
Compound 1 (100 mg, 9.6 μmol) was dissolved in a 1:1 mixture of CS2 and CH2Cl2 (30 ml). In a separate vial, TCNQ (10 mg, 5.2 μmol) was dissolved in a 1:1 mixture of CS2 and CH2Cl2 (3 ml). The two solutions were mixed thoroughly. Partial evaporation of the solvent resulted in the crystallization of 3, which was isolated in a 22.7% yield (30 mg, 1.2 μmol), mp decomposition 275 °C. Anal. calc. (found) for C51H4F24Hg6N4S6: C, 24.26 (23.78); H, 0.16 (0.15).
4.4 Crystal structures
X-ray data for 2 and 3 were collected on a Bruker SMART-CCD diffractometer using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). Specimens of suitable size and quality were selected and glued onto a glass fiber with superglue. The structure was solved by direct methods, which successfully located most of the non-hydrogen atoms. Subsequent refinement on F2 using the SHELXTL/PC package (version 5.1) allowed location of the remaining non-hydrogen atoms. Further crystallographic details can be found in Table 1 and in the Supporting Information.
Acknowledgments
Acknowledgment is made to the donors of the American Chemical Society Petroleum Research Fund for support of this research (Grant ACS PRF# 38143 -AC 3).
Supplementary material
Crystallographic data, atomic coordinates for compounds 2 and 3 have been deposited with the Cambridge Crystallographic Data Centre (CCDC), 12 Union Road, Cambridge CB2 1EZ, UK as deposition number CCDC 224188 & 224189, and can be obtained by contacting the CCDC.