

1 Introduction
The objective of this work was to study the effect of acoustic cavitation on actinide compounds. Acoustic cavitation is related to ultrasonically induced formation, pulsation and, under the proper conditions, implosive collapse of gas-filled micro-bubbles in liquids. The radius of the cavity immediately before the collapse, Rmax, can be estimated by the following equation [1]:
| (1) |
The energy release after the collapse also causes the heating of thin liquid shell surrounding the cavitating bubbles, where the sonochemical reactions of non-volatile species are also possible. According to the model developed in [1] the liquid reaction zone extends only approximately 200 nm from the bubble surface and has an effective lifetime of less than 2 μs after collapse. The size of the heated shell corresponds to a reactive liquid layer about 500 molecules thick. The temperature of the liquid reaction zone is much lower than that of the gaseous reaction zone of the cavitating bubble and estimated to be equal to several hundred Kelvin. Numerous examples of the sonochemical reactions in the liquid shell, like dissociation of nitrate ions or degradation of non-volatile organic and metallo-organic compounds [2,3], show that the rate of such reactions is much less than that of the volatile reagents. Sonochemical reactions in the interfacial region can be defined as the reactions occur under the ‘non-direct’ effect of acoustic cavitation.
Almost all recent studies of the sonochemical reactions of actinides were related to the ‘non-direct’ effect of cavitation in aqueous solutions [4–6]. So, sonolysis of actinide compounds in nitric acid solutions causes, first of all, nitric acid decomposition, followed by formation of nitrous acid. Sonochemically formed nitrous acid is the origin of U(IV), Np(V), and Pu(III) oxidation in nitric acid under ultrasound. At the same time, nitrous acid is able to reduce Np(VI) in sonicated nitric acid solutions. Recently, it was shown [7] that the sonochemical decomposition of hydrazine in nitric acid medium also is related to HNO2 sonochemical formation. Hydrogen peroxide formed in sonicated HNO3 in the presence of hydrazine causes U(IV) oxidation and Pu(IV) reduction [5,7,8].
It is evident that the chemical precursors of actinides should be volatile, in order to study the ‘direct’ effect of extreme conditions created by acoustic cavitation. Carbonyls of lanthanides and actinides are known to be unstable. However, the chelates of actinides with β-diketones can be suitable for studying such a ‘direct’ effect. Actinide(IV) β-diketonates are air-stable, hydrocarbon-soluble compounds that are easily prepared, purified and handled [9,10]. These compounds can be sublimated without decomposition at the temperatures of 150–250°C and reduced pressure. Sonolysis of some thorium(IV) β-diketonates have been studied recently [11]. It was found that the rate of sonolysis increases with the rising β-diketonate volatility. Solid sonication products consisted of a mixture of thorium dicarbide ThC2 and Th(IV) β-diketonates partial degradation products (PDP). Formation of ThC2 and PDP was attributed to the high-temperature reactions within the bubble and to the Th(IV) β-diketonates decomposition in the liquid reaction zone, recpectively. Kinetic study of sonolysis of some d and f transition-metal β-diketonates in hexadecane [12] confirmed that the sonochemical degradation of these compounds can be interpreted using a two-site model: metal β-diketonates with high vapour pressure are first decomposed in the gaseous phase of the cavitating bubble, and the sonolysis of less volatile complexes tends to occur in the liquid shell surrounding the bubble. In this work, we studied the sonolysis of some actinide(IV) β-diketonates, namely, Np(IV) and Pu(IV) complexes with hexafluoroacetylacetone (HFAA) and dibenzoylmethane (HDBM), respectively. Also some results were obtained on the composition of the solid sonication products for Th(IV) with HFAA and Pu(IV) with HDBM.
2 Experimental
Complexes of Th(IV), Np(IV), and Pu(IV) with β-diketones were prepared by mixing An(IV) solutions in 0.5 M HNO3 with the corresponding β-diketone solution in ethanol in the molar ratio of 1:4. The solids were then precipitated by adjusting the pH to 2–3 with 2.0 M CH3COONa solution. Solid complexes were obtained by the dissolution of precipitates in pentane followed by evaporation of the solvent. Complex of U(IV) with HFAA was found to be oxidized during precipitation. Complex U(DBM)4 is stable in the solid phase. However, it is oxidized rapidly in hexadecane solutions. More detailed data on U(IV) β-diketonates synthesis and stability are presented in [12].
Hexadecane used as a solvent in sonochemical experiments was dried over metallic sodium. Concentration of water did not exceed 100 ppm in dried solvent according to analysis with the Karl Fisher method. All solvents and prepared complexes were stored in a desiccator over anhydrous silica gel.
Spectrophotometric measurements were performed using a Shimadzu UV-3100 spectrophotometer. Infrared spectra were recorded in KBr pellets with a Nicolet Magna-IR 550 Spectrometer. X-ray powder diffraction data were obtained with Cu Kα radiation (λ = 1.5418 Å).
The transmission electron micrographs (TEM) were obtained on a JEOL-JEM 100SX microscope, working at 80-kV acceleration voltage. Samples for TEM were prepared by placing a drop of the sample suspension in ethanol on a copper grid coated with a carbon film and allowing it to dry in air.
Sonochemical experiments were performed using two kinds of sonochemical equipments.
Sonoreactor 1. Complexes of Th(IV) and Np(IV) were sonicated in a thermostatically controlled glass batch reactor with a volume of 25 ml by means of an ultrasonic device with working frequency 22 kHz and maximum electric output power 88 W. The sonicator was equipped with a piezoelectric transducer and a titanium horn with an irradiative surface area S = 3.5 cm2. The horn was immersed reproducibly 4 cm below the surface of the sonicated liquid. The sonochemical reactor was sealed with a plastic cover and bubbled with argon (100 ml min–1) through a fine tube for 10 min before sonication. The acoustic power P(W ml–1) transmitted to the system was measured by thermal probe method [13]. The ultrasonic intensity I (W cm–2) was then calculated as the P S–1 ratio. Obtained P and I values were found to be equal to 0.42 W ml–1 and 3.0 W cm–2, respectively.
Sonoreactor 2. Sonolysis of Pu(DBM)4 was performed with a 20-kHz ultrasound from a titanium horn (S = 1 cm2) driven by a piezoceramic transducer VibraCell 600. The solution volume subjected to ultrasound was 80 ml at a constant temperature maintained by a Huber Unistat Tango thermo-cryostat. The sonochemical reactions were performed under an argon atmosphere similar to those of sonoreactor 1. The ultrasonic cell 2 is shown in Fig. 1. The values of P and I obtained with the thermal probe were found to be equal to 0.43 W ml–1 and 35 W cm–2, respectively. Thus, these two sonoreactors allow studying the sonochemical processes at quite different ultrasonic intensities, but at similar absorbed acoustic power, due to the different diameter of the titanium horn and to the volume of the sonicated solutions.

Ultrasonic cell 2. f = 20 kHz, S = 1 cm2, I = 35 W cm–2, V = 80 ml, P = 0.43 W ml–1.
3 Results and discussion
Sonolysis of metal complexes in hexadecane solutions causes a change in the absorption spectra and the formation of solid products. From the UV/VIS/NIR spectra of Np(HFAA)4 and Pu(DBM)4 presented in Fig. 2, it follows that the sonication causes the light absorption intensity to decrease at the selected wavelengths typical for An(IV) β-diketonates. Increased light absorption in the UV region of the spectrum of sonicated Np(HFAA)4 (Fig. 2a, 2) is, most probably, due to the appearance of colloidal products of sonolysis. As compared to the spectrum of Np(HFAA)4 before sonolysis (Fig. 2a, 1), the spectrum of the sonicated solution of Np(HFAA)4 contains the new broad peak at 1005 nm, which can be related to the complex formation between Np(IV) and degradation products of β-diketones in hexadecane. The typical kinetic curves of Np(HFAA)4 and Pu(DBM)4 sonolysis are shown in Fig. 3. Their linearity in the semi logarithmic scale indicates the first-order kinetic law with respect to metal β-diketonate concentrations. The calculated values of the first-order rate constant, kobs, are presented in Table 1. The rate constants of sonolysis for Np(HFAA)4 and Pu(DBM)4 are similar to those for Th(HFAA)4 and Th(DBM)4 respectively. Volatilities of metal hexafluoroacetylacetonates are similar for all studied An(IV) as well as the volatilities of metal dibenzoylmethanates are also close to each other for An(IV). Thus, the data obtained confirmed that kinetics of metal β-diketonates sonolysis is related to the metal complexes vapour pressure. In the case of the more volatile complexes with HFAA [9,10], the sonochemical decomposition tends to occur in the gaseous phase of the cavitating bubbles. The sonolysis of the less volatile complexes with HDBM first occurs in the liquid shell around the cavitating bubbles. As the gas-phase reaction zone is the dominant site of the sonochemical reactions, the overall sonolysis rate is higher for An(HFAA)4 than for An(DBM)4. Rate constants for Pu(DBM)4 and Th(DBM)4 are the similar, although the ultrasonic intensities were quite different in these experiments and equal to 35 and 3.0 W cm–2, respectively. At the same time, the absorbed acoustic powers were approximately the same and equal to 0.43 and 0.42 W ml–1 for Pu(DBM)4 and Th(DBM)4, respectively. One can conclude that acoustic power absorbed by solution plays a more important role in the kinetics of sonochemical reactions than the ultrasound intensity under the experimental conditions used here.
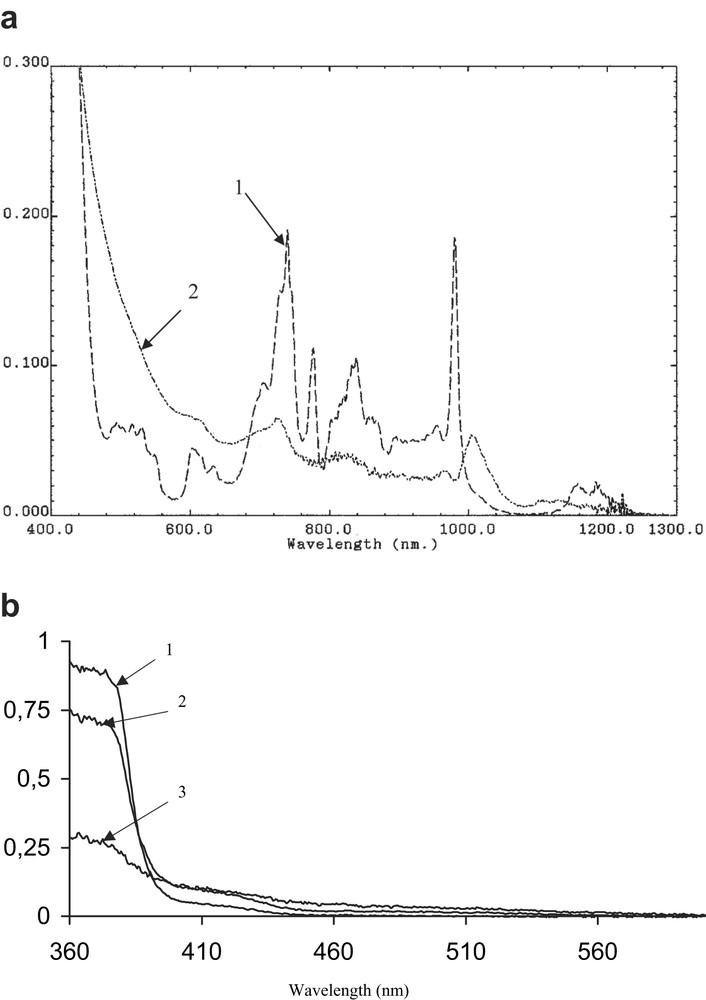
UV/VIS spectra of sonicated actinide(IV) β-diketonates. (a) Np(HFAA)4 (1.5 × 10–3 M) before (1) and after 180 min (2) of sonication (I = 3.0 W cm–2, Tson = 92 °C, P = 0.42 W ml–1, hexadecane, Ar). (b) Pu(DBM)4 (1.0 × 10–3 M) before (1) and after 60 min (2), and 240 min (3) of sonication (I = 35 W cm–2, V = 80 ml, P = 0.43 W ml–1, Tson = 103 °C, hexadecane, Ar).
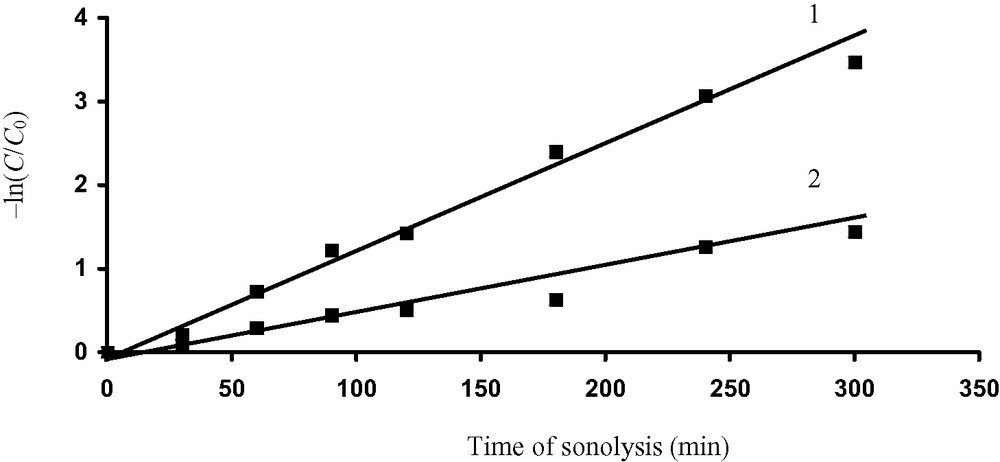
Kinetic curves of actinide(IV) β-diketonates sonolysis in hexadecane in the presence of Ar. (1) Np(HFAA)4, I = 3.0 W cm–2, P = 0.42 W ml–1, Tson = 92 °C; (2) Pu(DBM)4, I = 35 W cm–2, P = 0.43 W ml–1, Tson = 103 °C.
Observed first-order rate constant, kobs, for An(IV) β-diketonates sonolysis in hexadecane solutions. f = 20 kHz, Ar atmosphere
| Complex | kobs × 102 (min–1 W–1 ml) | Conditions |
| Np(HFAA)4 | 2.6 ± 0.4 | Sonoreactor 1, T = 92°C |
| Th(HFAA)4a | 2.1 ± 0.3 | Sonoreactor 1, T = 92°C |
| Pu(DBM)4 | 1.1 ± 0.1 | Sonoreactor 2, T = 103°C |
| Th(DBM)4a | 0.9 ± 0.1 | Sonoreactor 1, T = 92°C |
a Data [11].
Any solids were found to be formed under the effect of power ultrasound on neat hexadecane. By contrast, sonolysis of An(IV) β-diketonates in hexadecane solutions results in the formation of solid products. Solids were removed from solutions by centrifugation, washed twice with pentane and dried in an argon flow. Th(HFAA)4, Np(HFAA)4, and Pu(DBM)4 give grey, grey–green and black–brown products, respectively. FTIR spectra of Pu(DBM)4 and its sonication products are presented in Fig. 4a and b, respectively. The assignments of observed frequencies in the FTIR spectra of initial Pu(DBM)4 and its sonication products are presented in Table 2. The FTIR spectra were assigned using published data on vibration spectra of metal β-diketonates [14]. In general, FTIR spectrum of Pu(DBM)4 sonication product confirms important structural modifications of metal β-diketonates during sonication, observed recently for Th(IV) β-diketonates [11]. Several conclusions may be proposed concerning the mechanism of Pu(DBM)4 sonolysis from the comparative analysis of FTIR spectra:
- • (1) the disappearance of the ν(C–H)ar ,ν(C–C6H5), and ν(CH) vibration bands indicate that C6H5 groups are eliminated from the ligand molecules during sonication;
- • the disappearance of the ring deformation band intensity shows that sonolysis of Pu(DBM)4 causes the rupture of the chelate rings;
- • the characteristic new bands related to carboxylate νas(COO) and ν(C–H) typical for p-substituted phenyls demonstrate that the solid sonication product, most probably, contains carboxylate complexes of plutonium formed after Pu(DBM)4 destruction;
- • absorption band at 1384 cm–1 in the FTIR spectrum of the sonication product is superposed with another one centred at lower frequencies (≈1360–1350 cm–1); this absorption band can probably be related to C=C2– group vibrations in metal carbides [14]. A new absorption band in the FTIR spectrum of the sonication product at 669 cm–1 can be attributed to Pu–C stretch vibrations in metal carbide. Formation of plutonium carbides during Pu(DBM)4 sonolysis can be assumed by resemblance with sonolysis of Th(IV) β-diketonates where formation of ThC2 was observed [11].
| (2) |
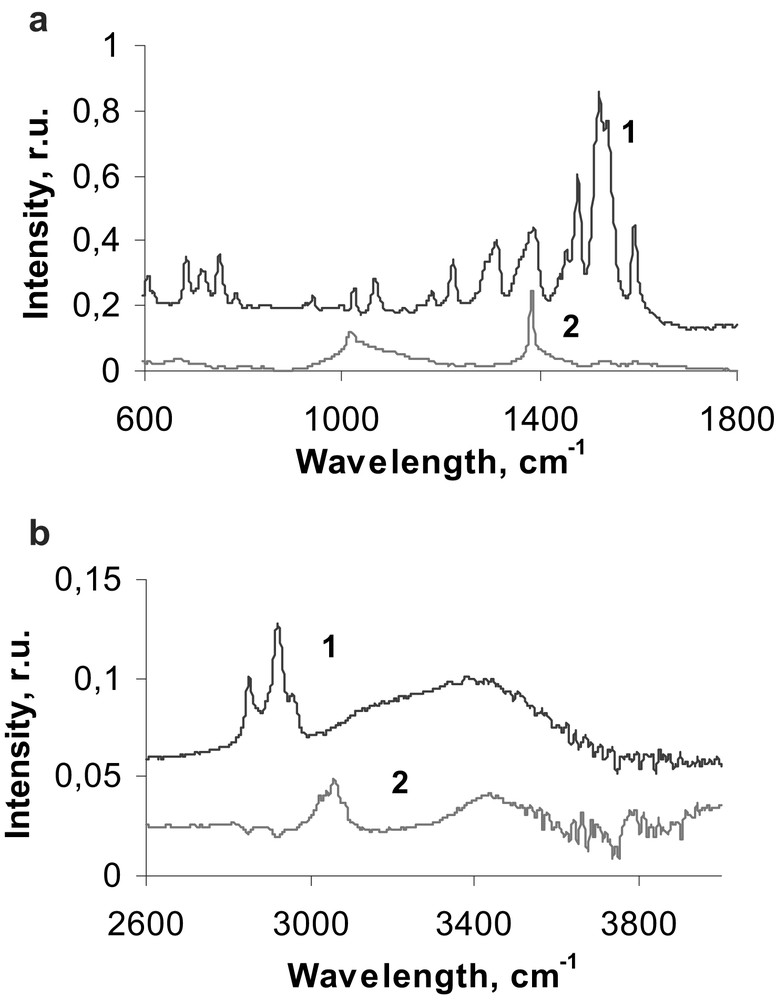
FTIR spectra of Pu(DBM)4 (1) and of its sonication product (2).
FTIR absorption peaks and assignments for initial Pu(DBM)4 and sonication products
| Frequency N (cm–1) / Intensity (a.u.) | Assignment | |
| Pu(DBM)4 | Sonication product | |
| 3057/weak | not observed | ν(C–H) aromatic |
| not observed | 2921–2851/medium | ν(C–H) aliphatic |
| 1592/medium | 1594/weak | ν(C=C); ν8 |
| 1536–1521/strong | 1537/weak | ν(C=O); ν1 |
| 1477–1455/medium | not observed | ν(C=O) + ν(CH); ν9 |
| 1388–1313/medium | very weak bands at 1400-1390 cm–1 overlapped with the strong band at 1384 cm–1 | ν(C=C) + ν(C–C6H5); ν2 |
| not observed | 1384/strong | νs(COO) in carboxylates |
| 1225/medium | not observed | ν(C-H); ν10 |
| not observed | 1160/weak | ν(C=C2–) in metal carbides (?) |
| not observed | 1019/mediuma | ν(C–H) aromatic in p-substituted phenyls |
| 751–715/weak | not observed | ν(C–H) |
| 685/weak | not observed | Chelate ring deformation; ν4 |
| not observed | 669/weak | ν(Pu–C) (?) |
| 606/weak | not observed | ν(C-C6H5)+ν(Pu-O); ν12 |
a Several weak bands at 1030–1050 cm–1 are overlapped with more intense band at 1019 cm–1
Comparative XRD data for annealed products of Th(HFAA)2 sonolysis and conventional ThO2 or ThC2
| Th(HFAA)4 products | Sonication products | ThO2 [15] | ThC2 [16] | ||||
| Heated at 800 °C in Ar | Heated at 800 °C in vacuum | ||||||
| dexp (Å) | I | dexp (Å) | I | dhkl (Å) | I | dhkl (Å) | I |
| 3.531 | weak | 3.538 | weak | ||||
| 3.289 | strong | 3.297 | very strong | ||||
| 3.258 | weak | 3.269 | strong | ||||
| 3.228 | strong | 3.231 | medium | 3.236 | very strong | ||
| 2.867* | weak | 2.954 | strong | ||||
| 2.795 | medium | 2.801 | medium | ||||
| 2.597 | very weak | 2.642 | medium | ||||
| 2.575 | very weak | ||||||
| 2.258 | very weak | ||||||
| 2.110 | weak | ||||||
| 2.086 | very weak | 2.068 | medium | ||||
| 2.008 | weak | ||||||
| 1.978 | medium | 1.980 | strong | ||||
| 1.975 | medium | ||||||
| 1.925 | weak | 1.964 | medium | ||||
| 1.681 | medium | 1.689 | strong |
The low-resolution TEM pictures of as-prepared and annealed materials are shown in Fig. 5. As-prepared material consists of very small particles (d ≤ 10 nm) with low extent of crystallisation. Heating in vacuum causes formation of nanocrystalline particles having tetragonal or round-shape symmetry. The average particles size is about 20–30 nm.


Low-resolution TEM pictures of as-prepared product obtained after Th(HFAA)4 sonolysis (a) and as-prepared product heated in vacuum at 800 °C (b).
The mechanism of actinide carbides formation under power ultrasound cannot be related to only thermal decomposition of An(IV) β-diketonates, since it is known that thermolysis of lanthanide or actinide β-diketonates causes corresponding metal oxides to form even in inert atmosphere or vacuum [18]. Conventionally, actinide carbides can be prepared by carbothermic reduction of corresponding actinide oxides with carbon or hydrocarbons at temperatures exceeding 1500 °C [17]. Therefore, it looks improbable that actinide carbides are formed in the liquid reaction zone surrounding the cavitating bubbles so far as the peak effective temperatures reached in a liquid zone is insufficient to obtain carbides. It can be assumed that the sonochemical mechanism of actinide carbides formation includes some high-temperature processes within cavitating bubbles. Under our experimental conditions, the cavitating bubbles contain argon, hexadecane vapour and An(IV) β-diketonate vapour. Sonolysis of alkanes is known to result in the formation of H2, C2H4, and C2H2 as primary products, with smaller amounts of C3 through C9 1-alkenes and alkanes [19]. The Rice radical-chain mechanism, which was originally demonstrated for alkane high-temperature pyrolysis, is consistent with the product distribution of alkane sonolysis. Also the light emission from C2* (d3πg → a3πu) excited molecules was observed in the multibubbe sonoluminescence spectra of sonicated alkanes [20]. The effective emission vibrational temperature of C2* particles was found to be equal to 4300 ± 200 K [21]. Thus actinide carbides formation inside the cavitating bubble could be due to the reactions of radicals formed from hexadecane sonochemical fragmentation and actinide(IV) β-diketonates or their degradation products :
| (3) |
| (4) |
| (5) |
| (6) |
Formation of PDP of An(IV) β-diketonates needs much lower temperatures that those for actinide carbides. Thus its formation can occur in the liquid reaction zone surrounding cavitating bubbles due to ‘non-direct’ effect of ultrasound. FTIR data show that PDP, probably, contain An(IV) carboxylates. This is in a good agreement with data on metal β-diketonates thermolysis published in [17], where it was shown that polymerised metal carboxylates are formed as intermediate products of metal β-diketonates thermal decomposition at approximately 300 °C in inert atmosphere. Thermal destruction of metal β-diketonates is accompanied by elimination of the side substitutes from the ligands and the rupture of the chelate rings in the complexes similar to those observed during metal β-diketonates sonolysis.
Finally, one may say that the composition of solid sonication products as well as the kinetic data indicate that actinide(IV) β-diketonates sonolysis occurs both in vapour and in liquid phases.
4 Conclusions
1. Tetrakis-β-diketonate complexes of actinide(IV) are decomposed under the effect of power ultrasound in alkane solutions. The mechanism of sonolysis with hexafluoroacetylacetone and dibenzoylmethane can be interpreted using a two-site model of the sonochemical processes: gaseous phase of the cavitating bubble and liquid shell surrounding the bubbles.
2. Sonolysis of more volatile complexes tend to occur within the cavitating bubbles. Transient high temperatures during acoustic collapse promote destruction of complex and solvent, followed by radical reactions with metal carbides formation as a final product.
3. Sonolysis of less volatile complexes first occur in the liquid reaction zone. Sonochemical decomposition of actinide(IV) β-diketonates in a liquid phase consisted of an elimination of the side substitutes from the ligands and the rupture of the chelate rings.
Acknowledgements
The authors thank Dr I. Charushnikova for the assistance in powder XRD analysis and Dr Yu. Koltypin for the help in TEM measurements. We are also grateful to Dr A. Maslennikov for his help in work with vacuum furnace. We gratefully acknowledge the support brought by the CE/DEN/DSOE and CEA/DEN/DDIN to this work.