

1 Introduction
Hydrogen-atom abstraction is a key step in numerous oxidation reactions in biology, in industrial processes or in the chemical laboratories. Hydrocarbon oxidations in industry include both homogenous and heterogeneous catalysis. The large use in organic synthesis of reagents such as Mn or Cr derivatives involves reactions that admit H-atom-abstraction steps. Homolytic C–H bond cleavage is formally a one-electron oxidation of the carbon atom and involves H-atom transfer (HAT). It is widely encountered as a key step in the catalytic cycle of oxidizing enzymes. An alternative mechanism is often proposed (electron plus proton abstraction or proton-coupled electron transfer (PCET), and the situation is often ambiguous. Nevertheless, the concerted mechanism HAT is thermodynamically favoured in the sense that it provides charge compensation that avoids charge separation in the reaction. The H-atom can exhibit tunnelling, which is experimentally detected by isotope effects. On the contrary, the non-concerted PCET implies either spatial or temporal resolution of the two elementary processes and the two components (H+, e–) may take distinct paths between donor and acceptor.
In this paper, we briefly summarize the chemical features of homolytic C–H bond cleavage and we focus attention on some selected enzymes (with a particular emphasis on galactose oxidase) presumably working according to this process.
2 Organic radicals promote homolytic C–H bond cleavage
2.1 Basic concepts: frontier-orbital approach [1] and thermodynamics of C–H bond oxidation [2]
Most of isolated radicals do not have a charge, and in most chemical reactions they react with uncharged molecules. Thus the coulombic forces are usually small while the frontier orbitals interactions are large. The frontier orbital of a radical is the singly occupied one (SOMO). This orbital can interact with both the HOMO and the LUMO of the substrate (Fig. 1). Radicals with high SOMO react faster with molecules baring low LUMO and radicals with low SOMO react faster with molecules having high HOMO. Consequently, radicals with low SOMO are electrophilic radicals, while radicals with high SOMO are considered as nucleophilic radicals.
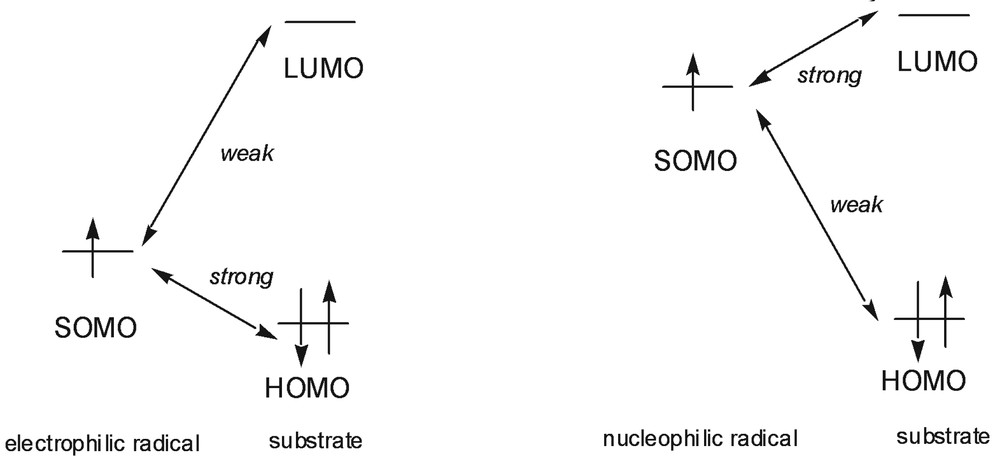
Frontier orbitals interactions for a radical.
When a radical attacks a C–H bond, the SOMO interacts with σ and σ* orbitals. The latter are high in energy and we can expect that the major interaction occurs with the HOMO (σ). Radical-abstracting hydrogen atoms are thus usually regarded as electrophilic (Fig. 2). Consequently, these radicals interact in the transition state more strongly with electron-rich hydrogen than with an electron-poor one. Oxygen radicals such as hydroxyl radical, alkoxyl radicals or aryloxyl radicals frequently encountered in biological processes belong to this class, while carbon radicals are nucleophilic one.
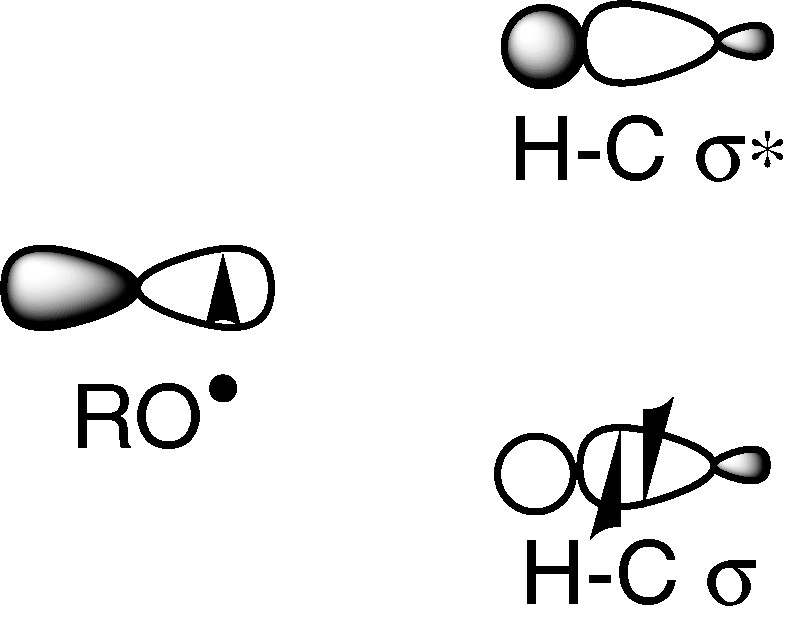
An electrophilic radical and a C–H bond.
Owing to the fact that total oxidation of organic molecules (combustion) is largely exothermic, the challenge lies in stopping the oxidation at an intermediate product (e.g., such as methanol from methane, in which the first hydroxylation is the least exothermic of four two-electron steps). In the Mayer’s approach [2,3], devoted to H-atom abstraction by metal oxo complexes, the importance of the C–H bond strength is underlined (vide infra). This approach may be extended to H-atom abstraction by organic radicals.
2.2 H abstraction by nitroxide or imidoxyl radicals in organic synthesis
Oxygen or sulphur radicals are involved in the catalytic cycles of some enzymes and will be discussed in the following of the paper. Some organic syntheses involving oxidation reactions promoted by nitroxide or imidoxyl radicals have been described [4–11]. The bis(trifluoromethyl) nitroxide and the phtalimide N-oxyle radical (PINO) are able to promote the homolytic C–H bond cleavage in alkanes, leading to the carbon radical in the first step of the reaction (Fig. 3).
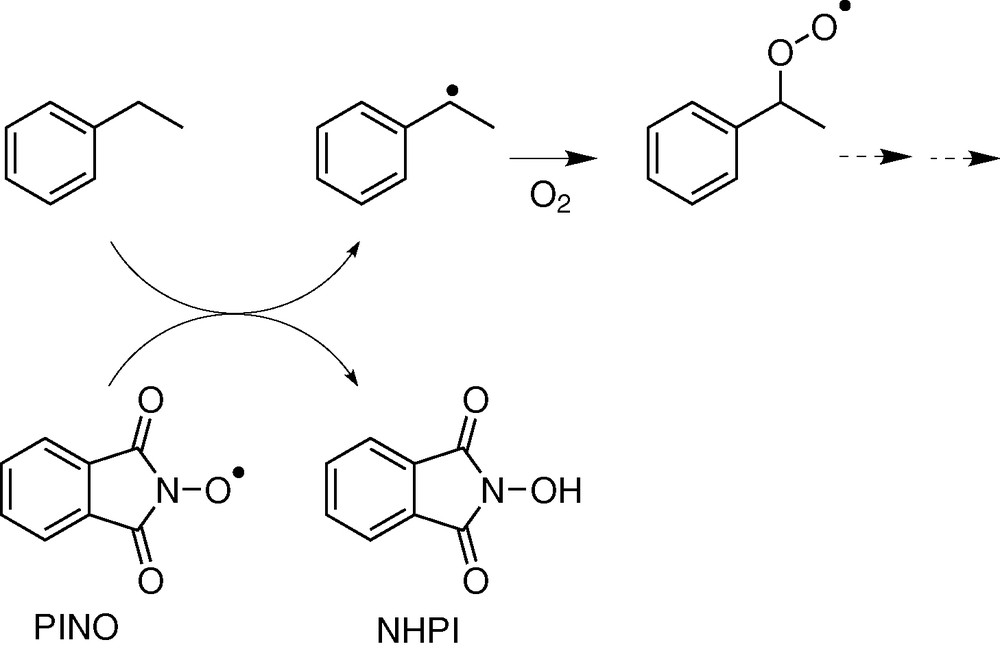
Hydrocarbon oxidation by PINO.
It has to be noted that nitroxide radicals, stabilized by the α-effect, possess high-energy SOMO (the unpaired electron is located in a π* level) and, consequently, have nucleophilic character. A cooxidant for the regeneration of the oxidizing radical is needed for a catalytic process.
3 H-atom abstraction by metal-oxo complexes
H-atom abstraction by metal-oxo complexes may involve the surface of the heterogeneous catalyst [12], some reagents in organic syntheses [13] and metalloenzymes.
Direct hydrogen abstraction by metal has been discussed by Shilov et al. [14,15]. A direct interaction of an alkane with a metal core is, as an example, involved by Shilov in the reaction of Pt(II) complexes with alkanes via oxidative addition:
The selectivity (primary C–H bond more reactive than the secondary one, more reactive than the tertiary one) turned out to be the opposite of that for radical reactions. These reactions do not involve metal-oxo species and are probably out of the scope of this paper.
3.1 Basic concepts: frontier orbitals for H abstraction by metal-oxo complexes [1] and thermodynamic approach [2]
The orbital interactions between a metal-oxo species and the frontier orbitals of a C–H bond are depicted in Fig. 4.
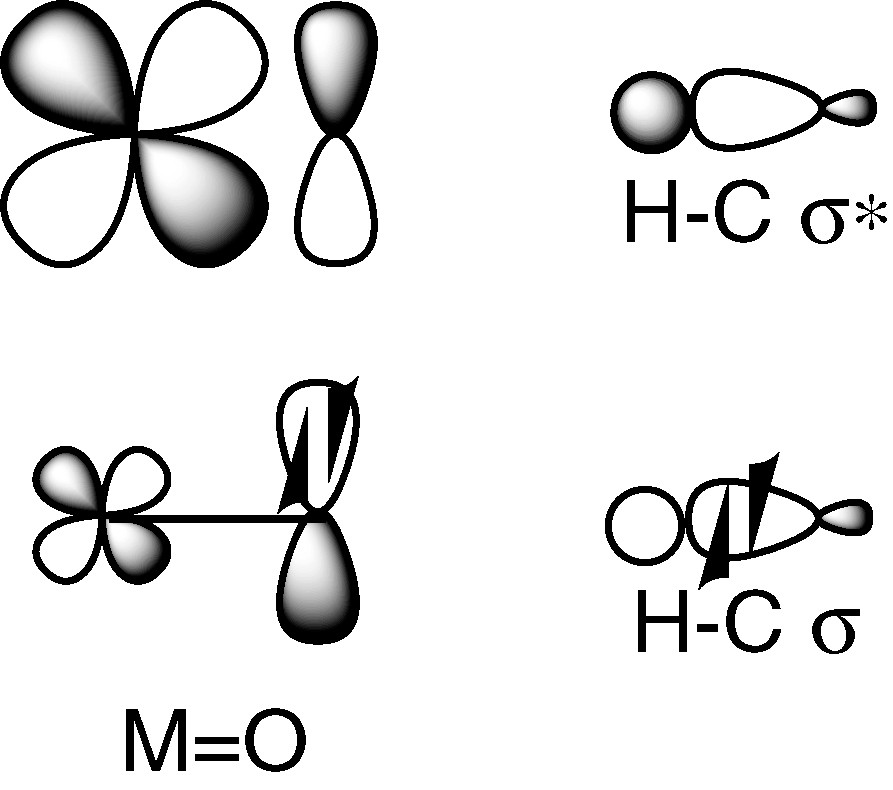
A metal-oxo complex and a C–H bond.
The situation is complicated owing to the fact that the HOMO and the LUMO of the oxidant are involved instead of only a SOMO, as in Fig. 2. Nevertheless, in two nice papers [2,3], J.M. Mayer underlines the analogy of H-atom abstraction by metal-oxo complexes and organic radicals reactions. An approach based on the thermochemistry of the H-atom transfer step is described and illustrated by the C–H bond oxidation by CrO2Cl2 and by MnO4–. The initial reaction of CrO2Cl2 with C–H (hydrocarbon) proceeds by H atom transfer to give a carbon radical:
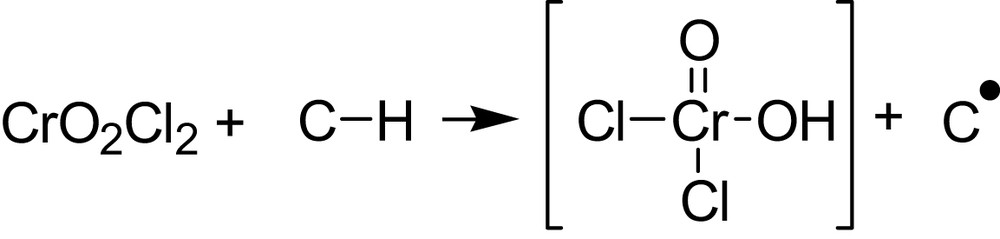
The rate of the initial H-atom transfer matches that of the disappearance of CrO2Cl2. The derived ΔH# and ΔG# for this step correlate well with the strengths of the C–H bonds being cleaved [2,3].
Permanganate oxidations of alkyl aromatic hydrocarbons seem also to involve transient carbon radicals, the rate constants correlating with C–H bond strengths. Although spin-density arguments would predict that CrO2Cl2 and MnO4– should not abstract H atoms, yet they do. The bond strength approach can give quantitative predictions of rate. A plot of log k vs the O–H bond strength of the abstractor (MnO4–, ROO•, RO•, HO•) shows that permanganate abstracts H• at essentially the rate expected for an oxygen radical that would make an 80-kcal mol–1 O–H bond. This occurs even though permanganate is not a radical. The work of J.M Mayer underlines that unpaired spin density is not required for H atom abstracting reactivity. The reactivity is routed in the strength of the bond the oxidant makes with H•. A high affinity for H• requires an oxidizing metal centre and the reduced form must have significant basicity. The importance of bond strength in H atom abstraction should be extended to coordination complexes, metal oxides active sites in heterogeneous catalysis, and active sites in metalloenzymes [2,3].
4 H-atom abstraction by metalloenzymes
We have only focused attention on the H-atom abstraction step; detailed introduction to the enzymatic systems and complete balance of the reaction are detailed in the cited references. It is often claimed that metalloenzymes that abstract H• have some ‘radical character’: M=O can be written with the resonance structure M–O• [16]. H-atom abstraction is a consensus step of iron-dependent systems belonging to different classes of metalloenzymes. The cytochrome P-450-dependent monooxygenases as well as the non-heme diferric methane monooxygenase (MMO) involve homolytic C–H bond cleavage of the substrate by a high-valent iron-oxo species that can be described as an electrophilic iron-oxyl radical to give an alkyl radical (R• in Fig. 5) [17–22]. Nevertheless, Mayer suggests that the radical character has little to do with H abstracting reactivity and that these reactions are rather controlled by the endothermicity of the H-atom transfer step.
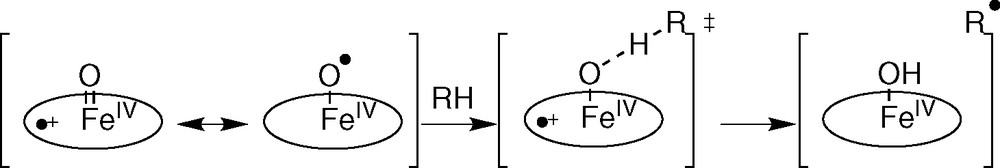
H abstraction by P 450 : a radical cage process.
Considerable evidence supports the existence of the carbon radical (R• in Fig. 5) that subsequently displaces the hydroxyl group from iron in a homolytic substitution (oxygen rebound mechanism): primary isotope effect, C–H bond selectivity, carbon skeletal rearrangements (ring opening, ring expansions, etc.) during oxidation of radical clock substrate probes. Nevertheless, the involvement of true radical intermediates has been questioned and a non-synchronous concerted mechanism was invoked [19–22].
Lipoxygenase [23–25] is a particular example of non-heme mononuclear iron enzymes in which a high-valent iron centre is not required for activation of the substrate, catalysis occurring through substrate activation rather than O2 activation. The mammalian enzymes act on arachidonic acid to produce hydroperoxides that are precursors of leucotrienes and lipoxins, mediators of inflammation. The catalytic centre contains a Fe(III)–OH species that is proposed to abstract the C-11(R) H atom of the substrate, leading to a carbon radical and a ferrous Fe(II)–OH2 centre (Fig. 6). A kinetic isotope effect indicates that C–H bond cleavage is involved in the rate-determining step. Kinetic experiments further confirm that the C–H bond cleavage occurs prior to the incorporation of O2 by the activated substrate-enzyme complex. A biomimetic model described by Stack et al. [23,24] reproduces this mechanism. The redox potential of the iron centre (600 mV/NHE) being too low for a pure outer sphere reaction (for 1, 4 cyclohexadiene E°1/2 = 1.1 V/NHE), only the approach of Mayer [26] can explain the observed reactivity: very weak strength (0.76 kcal mol–1) for the broken C–H bond, the strength of the formed O–H bond providing additional driving force for the reaction when compared to the purely outer sphere mechanism.
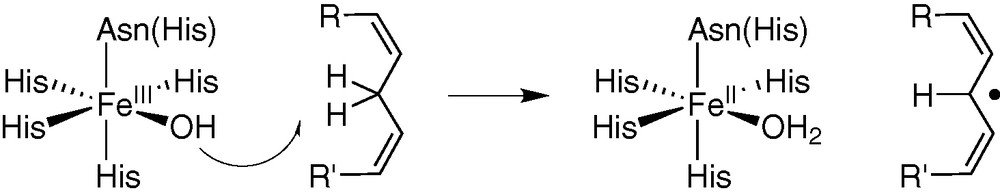
H-atom abstraction in lipoxygenase.
Similar process could arise with copper enzymes. Nevertheless, few copper enzymes are able of hydrocarbon C–H bond oxidation, owing to the impossibility to reach a highly oxidizing Cu(III) state (the ligand is oxidized before the metal). It has been proposed for dopamine β-hydroxylase (DBH) a homolytic C–H bond cleavage on the dopamine substrate by a transient Cu–O° species [27,28] (Fig. 7).
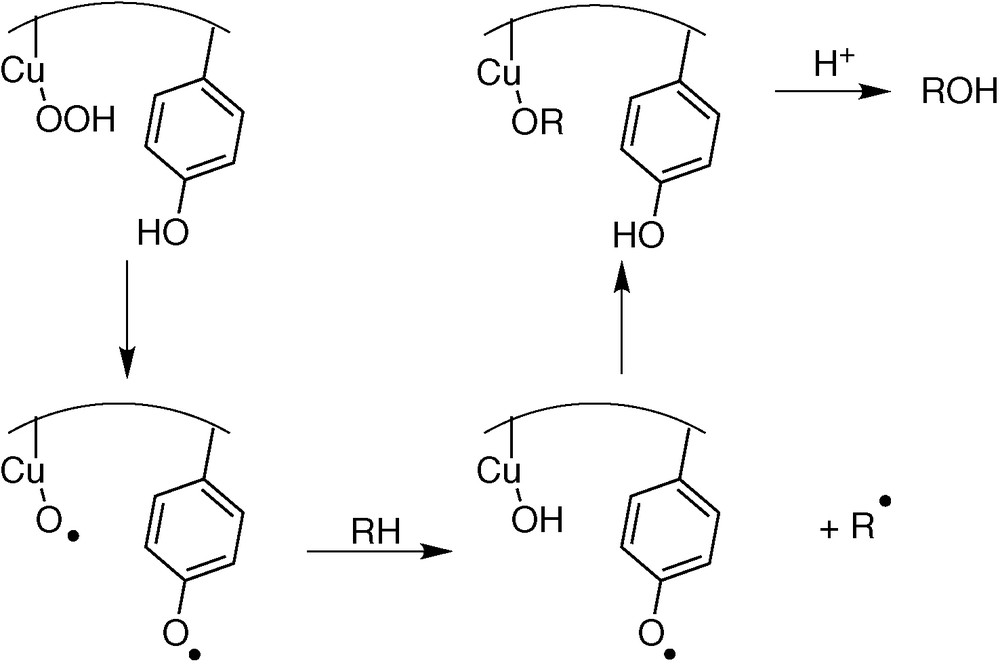
Proposed mechanism for DBH.
5 Radical enzymes
A fascinating class of metalloenzymes is that whose members utilize their own polypeptide chain as a cofactor and generate organic free radicals on specific amino acid residues [29–31]. The amino acids usually involved in such a redox process include tyrosine (class-I ribonucleotide reductase, photosystem II, prostaglandine H synthase, bovine liver catalase, DNA photolyase, galactose oxidase, glyoxal oxidase...), tryptophan (cytochrome c peroxidase) and glycine (ribonucleotide reductase, pyruvate formate lyase).
A homolytic C–H bond breaking is suggested on the basis of experiments and calculations in class I RNR [31]: a Cys 439 thiyl radical abstracts H-atom at C3' position of the substrate, leading to a carbon radical (Fig. 8).
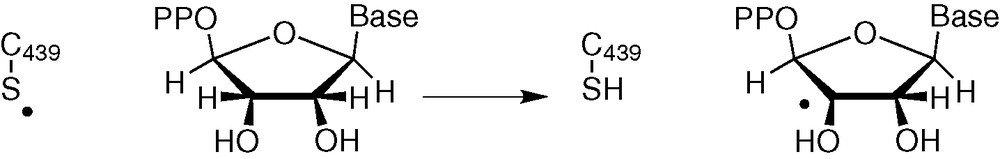
H abstraction by a thiyl radical (class I RNR).
A similar step is also suggested on the basis of B3LYP calculations for class-III RNR, the H abstraction being endothermic by 9.7 kcal mol–1, the formyl radical formed being stabilized by H-bonding [31]. Hydrogen atom abstraction by a thiyl radical (from a cysteine) is also observed with benzyl succinate synthase: a benzyl radical is created from a toluene methyl, this step occurring with a relatively low barrier of 12.9 kcal mol–1 [31].
In coenzyme B12-dependent enzymes, H-atom abstraction occurs from the substrate by an Ado-CH2• radical, leading to a carbon radical on the substrate [31] (Fig. 9). The barrier of the H-atom transfer has been calculated for substrate models and Ado-CH2• to be ca 10 kcal mol–1.
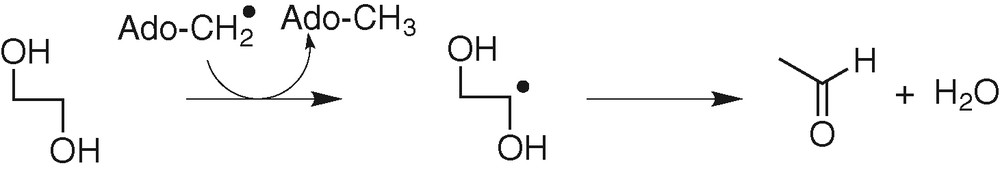
Example: diol deshydratase.
Some enzymes utilize a glycyl radical (pyruvate formate lyase, class-III anaerobic RNR, benzyl succinate synthase) to promote H-atom abstraction [31]. Nevertheless, looking to resonance structures of glycyl radicals, H-abstraction affording the radical may involve C–H, O–H or N–H homolytic bond breaking (Fig. 10).
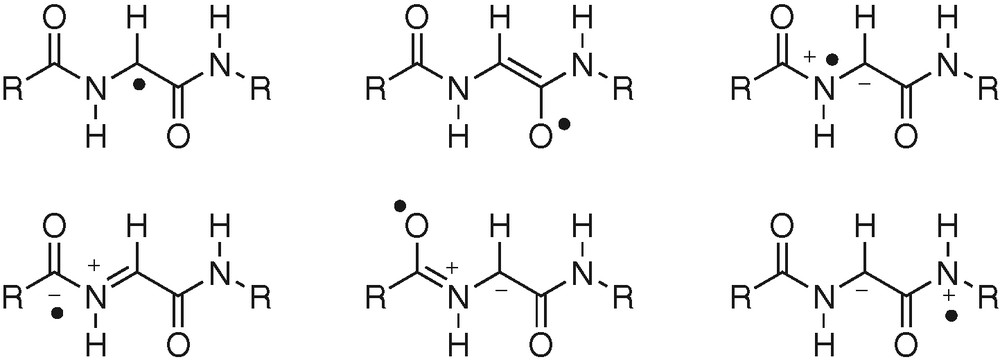
Resonance structures for glycyl radical.
Tyrosyl is the most frequently encountered radical in radical enzymes. As an example, we focus attention on the hydrogen-atom abstraction by prostaglandin synthase. In this enzyme, a tyrosyl radical is postulated to abstract an H atom from C13 of arachidonic acid to generate a protein-bound arachidonyl radical, which further initiates a cascade of radical reactions. The H-atom abstraction is stereospecific (only the pro-S hydrogen is abstracted) and this step is rate limiting (calc. 12.3 kcal mol–1; exp. 14.5 kcal mol–1) [31]. Fig. 11 depicts the initial attack by the tyrosyl radical and the optimised transition-state structure according to [31].
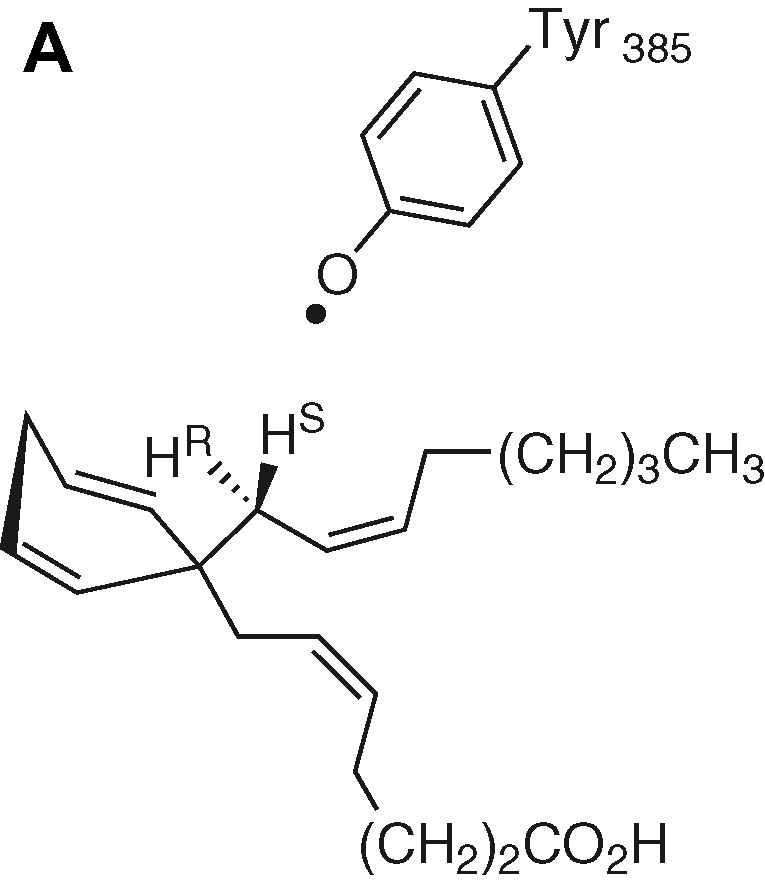
Abstraction of the pro-S H-atom and the optimised transition-state structure.
6 Galactose oxidase: the paradigm of a metal centre coupled to a tyrosyl radical
Galactose oxidase (GOase) is an extracellular type-II mononuclear copper protein (68 kDa) of fungal origin [32,33]. GOase catalyses the two-electron oxidation of a wide range of primary alcohols to their corresponding aldehydes with the concomitant reduction of molecular oxygen to hydrogen peroxide. The X-ray crystal structure [34] indicates that the copper has a square-based pyramidal geometry, with Tyr495 occupying the axial position. His 581, His496, Tyr272 and an exogenous ligand (H2O or acetate, replacing the substrate) coordinate the copper in equatorial position. An unusual feature is the cross-linking of Tyr272 to Cys228 through a thioether bond at the ortho position of the OH group, presumably to lower the tyrosyl/tyrosine redox potential. The indole ring of Trp290 is π-stacked with Tyr272 and controls the access to the active site. The enzyme can exist in three well-defined oxidation levels: the EPR-silent oxidized form (cupric ion antiferromagnetically coupled to the tyrosyl radical), an intermediate inactive copper(II)–tyrosinate form and the reduced copper(I)-tyrosine form. The proposed catalytic mechanism for GOase is presented in Fig. 12.
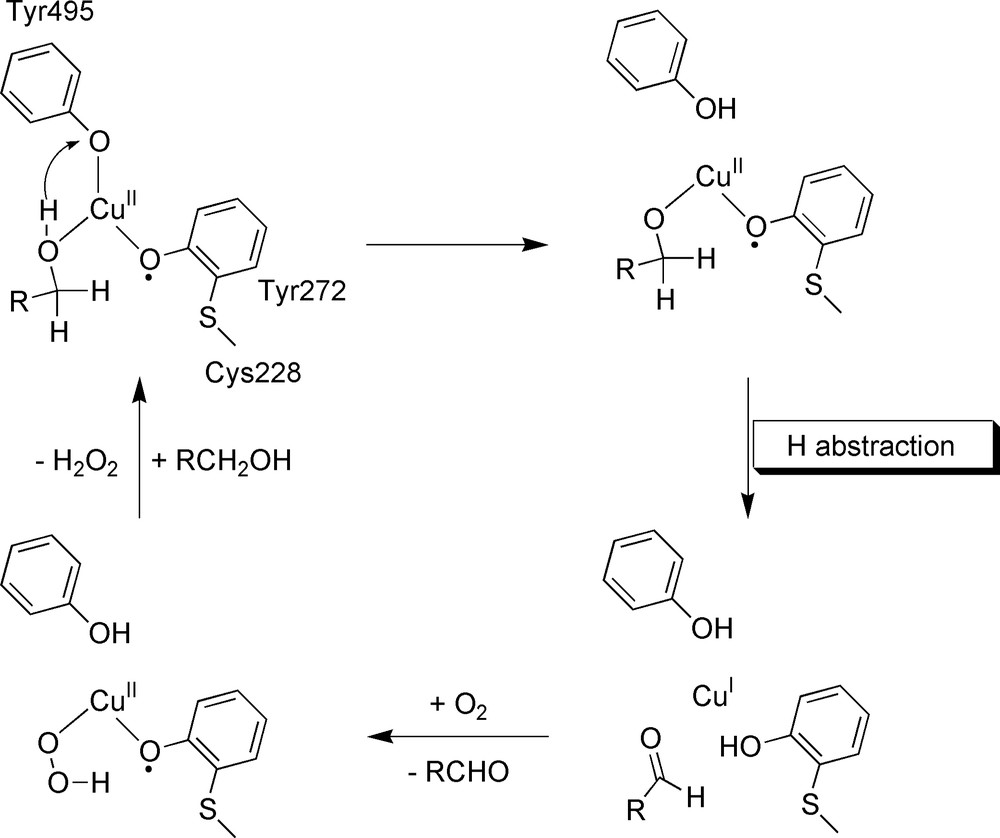
The catalytic mechanism in GOase.
In the ping-pong turnover reaction, now well established, the alcohol binds to the oxidized CuII–radical active site and reduces both redox centres. This reaction implies the breaking of a Cα–H bond of the substrate. In the second half reaction, the CuI–tyrosine site binds O2 and reduces it to H2O2, regenerating the initial CuII–radical state.
The catalysis is initiated by weak (almost collisional) association with the substrate. The reaction has been extensively studied with pyranoside substrates to avoid anomerism problems. Reaction over a wide range of concentrations shows that the enzyme is saturable with a dissociation constant close to 200 mM for the formation of the initial complexes; this low affinity, which is a consequence of a quite open active site, is consistent with a lack of substrate selectivity making GOase an excellent tool for studying transition state structures. In contrast, the orientation of the substrate is consistent with a stereospecific abstraction of the pro-S hydrogen by the nearby tyrosyl radical. After binding of the substrate to the copper equatorial position, the first step involves a proton transfer (PT): deprotonation of the alcohol is facilitated by copper coordination and a short H–OTyr495 bond (estimated to be ca 2.8 Å). This process occurs with a low barrier (nearly isothermic), within the ns timescale, due to the optimised hydrogen bonding geometry and the close matching of tyrosine pKa and coordinated-alcohol pKa. Two crucial facts result from this first step: the CuII–substrate interaction is strengthened and the C–H bond energy of the deprotonated substrate is lowered by 10 kcal mol–1. The second step involves a hydrogen-atom transfer (HAT) from the alkoxide substrate to the equatorial Tyr272 radical. It constitutes the major rate-limiting step (at high O2 concentrations) in the whole catalytic pathway, as reflected by (i) an unusually large kinetic isotope effect (KIE) (reaching 22 for 1-O-methyl-α-d-dalactopyranoside) and (ii) the fact that GOase remains essentially fully oxidized during turnover. However, if at low substrate concentrations the KIE is maximal, at higher substrate concentrations (at Km or higher), it dramatically decreases to 8, suggesting that there is also a primary KIE on the oxygen reduction half reaction. The relative contribution of each process consequently depends on dioxygen and substrate concentration but, under physiological conditions, substrate oxidation remains rate limiting in turnover. The large non-classical KIE of 22 as well as a strong temperature dependence of the KIE are clear evidences of the tunnelling of a hydrogen atom in the rate-limiting step. Transient reduction experiments at low substrate concentrations allowed the determination of an enzyme reduction rate constant of 1.5 × 104 M–1 s–1 at 4 °C (KIE of 22); the active site re-oxidation rate constant has been fitted to 8 × 106 M–1 s–1 (KIE of 8). The rate constants range between 177 and 2.7 × 104 M–1 s–1 at 25°C, depending on the primary alcohol, with a 9300 M–1 s–1 value reported for d-galactose.
The radical mechanism for oxidation of the substrate implies both an electron and hydrogen-atom transfer. These elementary processes do not occur in a concerted fashion, since the two redox centres are distinct: the inner sphere electron transfer to the CuII centre could follow or precede the HAT to the tyrosyl radical (Fig. 13). This ordering has been discussed in a recent paper by Whittaker et al. [33]. Quantitative structure–activity relationship correlations over the steady-state rate data for the oxidation of benzyl alcohol derivatives revealed that the three elementary events, namely PT, single electron transfer (SET) and HAT represent asynchronous components of the whole catalytic process, rather than distinct kinetic intermediates. The hardly oxidisable nitro derivatives as well as pyranoside and physiological substrates exhibit a large KIE, associated with a negligible solvent kinetic isotope effect (SKIE): The C–H bond cleavage (HAT) is here fully rate limiting. In contrast, the easier to oxidize methoxybenzyl alcohols exhibit a smaller KIE and a non-negligible SKIE, reflecting a small PT contribution in the TS and a pronounced SET character for the substrate oxidation. This is interpreted by a shift in the character of the TS: while ΔG‡ remains similar for both substrates, the TS for the oxidation of the methoxy derivatives (larger driving force) occur earlier along the reaction coordinate than oxidation of the nitro derivatives.

Hydrogen atom transfer vs single electron transfer for the substrate oxidation.
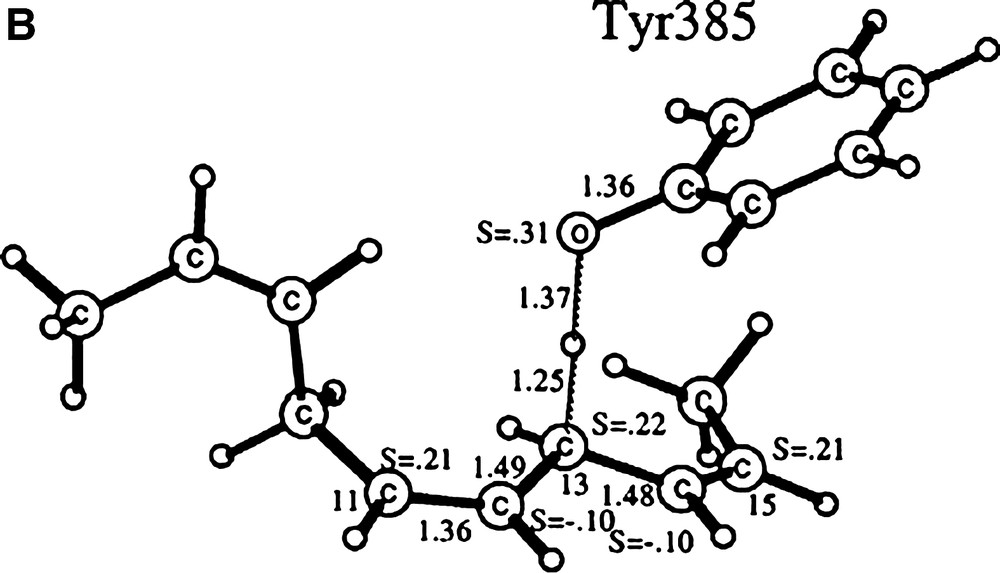
The free activation energy for the hydrogen abstraction has been estimated to 13.6 kcal mol–1. From B3LYP calculations, an optimised structure for the TS could be obtained: the O–H bond length is 1.24 Å, while the C–H bond length stretches to 1.36 Å. This five-atom cyclic TS, whose existence has been further confirmed by kinetic studies on the enzyme, brings the phenoxyl O near the H atom. The hydrogen atom is then transferred perpendicularly to the phenol ring plane, in agreement with a π character of the radical. Mechanism-based inactivation of GOase by the ultra fast radical probe trans-2-phenylcyclopropylmethanol confirms that a short-lived substrate ketyl radical anion results from the H abstraction (HAT is rate limiting). The ketyl radical then reduces the metal centre and is released to allow O2 binding.
Several model complexes for the active site of GOase have been synthesized in the last decade [35,36]. The structure of a complex bearing coordinated alcohol/alkoxide has been described and can be used to model the substrate binding prior to H abstraction. It was obtained by Tolman et al. [37] from the copper complex of a ligand derived from a phenolate tetraazayclononane backbone (N3O coordination sphere). The benzyl alkoxide ligand and the phenolate group are coordinated in a cis-equatorial fashion, with a 2.995-Å distance between one benzylic hydrogen and the phenolate oxygen. This structure reproduces the disposition of substrate and Tyr272 in GOase. Interestingly, this copper complex is active toward alcohol oxidation: its one-electron electrochemical oxidation at the phenolate/phenoxyl couple results in the bleaching of both the UV/Visible and the EPR spectra, consistent with the formation of a CuI phenol species. Analysis of the reaction mixture indicates formation of benzaldehyde. Catalytic primary alcohol oxidations by fully characterized CuII-phenoxyl radical species have been reported latter for few complexes [35,36]. As an example, 300 turnovers 1–1 d–1 were achieved for the aerobic oxidation of benzyl alcohol into aldehyde by the copper complex depicted in Fig. 14 [38]. The formation of H2O2 has been evidenced. The catalytically active one-electron oxidized species has been identified as the copperII-coupled phenoxyl radical located on the equatorial phenolate.
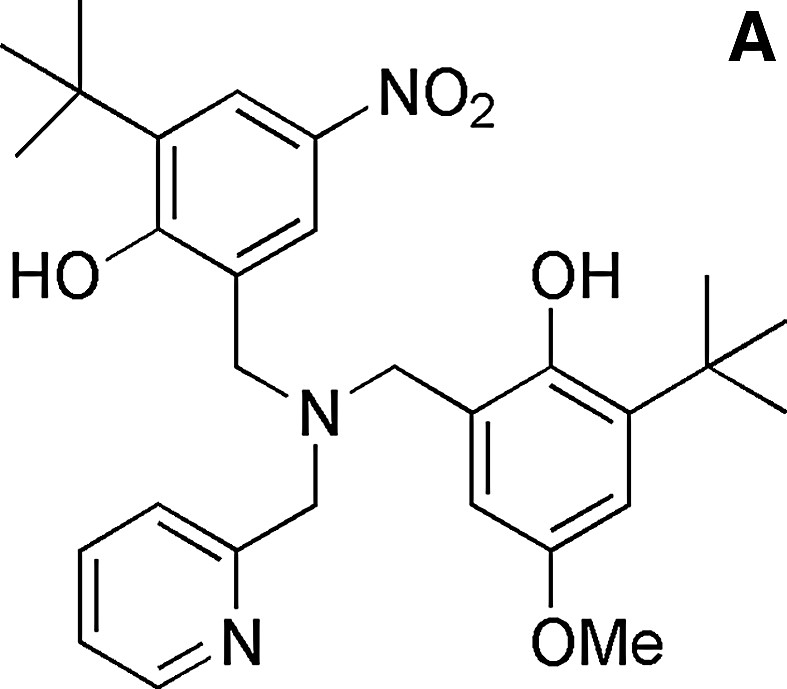
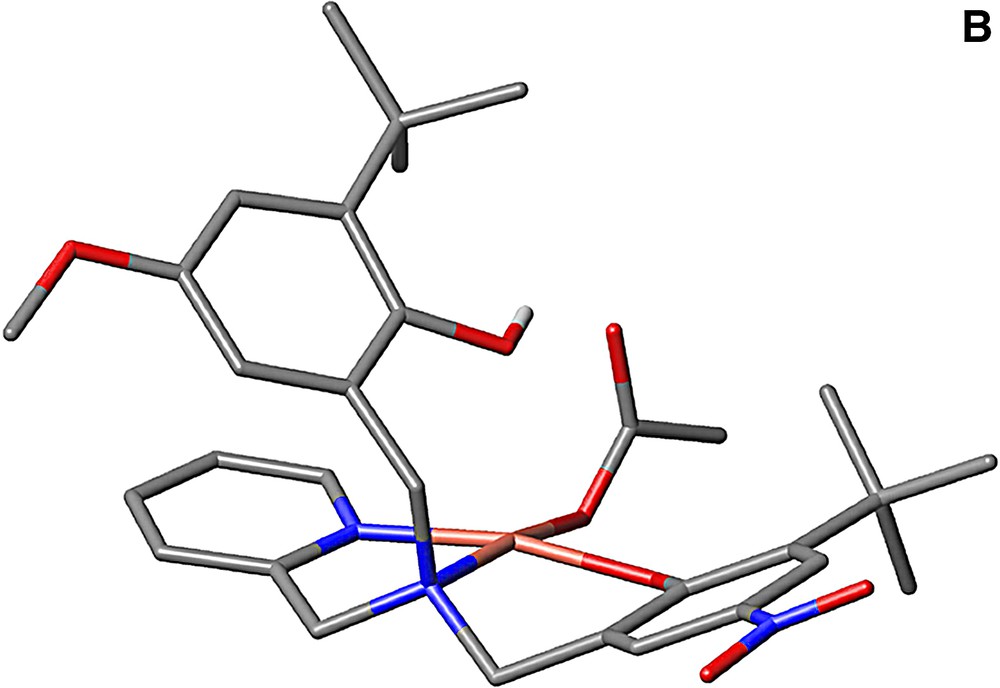
The ligand LH2 and its copperII complex (acetate).
Under single turnover conditions, kcu was estimated at 0.0271 M–1 s–1 and once again H abstraction was found (from isotopic effect) to be the rate-limiting step. Using unactivated primary alcohols, 40 turnovers were achieved for the oxidation of n-pentyl alcohol, 80 for ethanol. In addition, this model not only reproduces the redox chemistry of GOase, but also its chemoselectivity: using cyclohexanol or 2-hexanol, no trace of ketone could be detected, even after 1-week reaction.
7 Conclusion
Three primary factors affect the driving force for a H-atom abstraction:
- • the redox potential of the reaction centre (radical, metal);
- • the strength of the C–H bond broken;
- • the energy of the H–Y bond formed in the reduced reagent.
There are two possibilities in the mechanism of H-atom transfer to an organic oxidizing radical: a one-step hydrogen atom transfer or an electron transfer followed by proton transfer. The two mechanisms can be differentiated from the primary kinetic isotope effect in the case of phenoxyl radical-metal complexes. For a one-step H-atom transfer reaction, the kH2/kD2 value should decrease with increasing the binding strength of metal ions to the phenoxyl radical, while this value should increase in the two-step mechanism [39].
It can be emphasized that in metalloenzymes (as well as in biomimetic models) that catalyse oxidation processes, two main strategies are encountered. In the first one, oxygen is needed to generate the oxidizing species; it is the case of iron-oxo based systems. In the second one, oxygen is needed to regenerate the oxidizing species (catalytic process) but stoichiometric reaction is possible under anaerobiosis; it is usually the case of copper enzymes. A third strategy uses organic radicals i.e. non-metallic redox centres, working or not synergistically with metal centres to realize one electron at a time two-electron oxidations of organic substrates. The homolytic C–H bond cleavage is the key step of these reactions. A fascinating area is opened up to the imagination of chemists by the design of molecular species containing two and more oxidizing equivalents in an unique reagent involving metallic and non-metallic redox centres held together in a manner allowing a finely tuned cooperativity.
Acknowledgments
The authors are indebted to Prof. Éric Saint-Aman for helpful discussions and advice.