

1 Introduction
Emission from (L) Re (CO)3(α-diimine) complexes (L = axial ligand = halide, alkyl, benzyl, metal fragment) often provides the first indication of the character of the low-lying excited states. Moreover, detailed experimental studies of emission spectroscopy have played a central role in the understanding of their photochemical reactivity and photophysical properties [1]. This class of molecules is characterized by significant electronic delocalization effects on the three chemical centers determined by the metal atom, the α-diimine group and the ligand L [2]. Depending on these chemical centers irradiation into the visible may lead to a variety of important applications such as ultra-fast dissociation of a carbonyl ligand or homolytic cleavage of the Re–L bond with formation of di-radicals initiators of polymerization [3]. The molecules may also behave as strong luminophore or as precursors of electron/energy transfer.
The photoreactivity and excited states dynamics of this class of molecules have been investigated in details by means of accurate quantum chemical calculations completed by wave packet propagations for the HM(CO)3(H-dab) complexes (M = Mn, Re) [4,5]. Whereas a complete and ultra-fast direct dissociation of the CO ligand (in less than 500 fs) has been put in evidence by the theoretical study for the manganese complex a rather slow (beyond the ps time scale), non total and indirect homolysis of the Re–H bond occurs through 1MLCT → 3SBLCT intersystem crossing in HRe(CO)3(H-dab). This later reaction may certainly compete with the emission process either from the 1MLCT absorbing state or from the low-lying 3MLCT states, according to the schematic view of the potential energy curves (Scheme 1).

This is corroborated by the experimental quantum yield of the Re–L bond homolysis which may vary between 10–2 (L = methyl) to nearly 1.0 (L = ethyl or benzyl) [6,7] in these molecules.
The purpose of the present theoretical investigation is to simulate the absorption/emission spectra of HRe(CO)3(H-dab) by means of wave packet propagation on the one-dimensional potentials V(q) (q = [Re–H]) calculated for the MLCT (5dRe → π*H-dab) and the electronic ground state. The low-lying 3SBLCT and 3MLCT states are not included in this preliminary study.
2 Computational details
The potential energy curves corresponding to the electronic ground state 1A' (σRe–H)2(5dx2–y2)2(5dyz)2(5dxz)2) and low-lying 1MLCT absorbing state (5dRe → π*CO) were obtained by calculating the potential energy as a function of the Re–H bond length, keeping all other coordinates frozen at the idealized geometry depicted in Fig. 1.
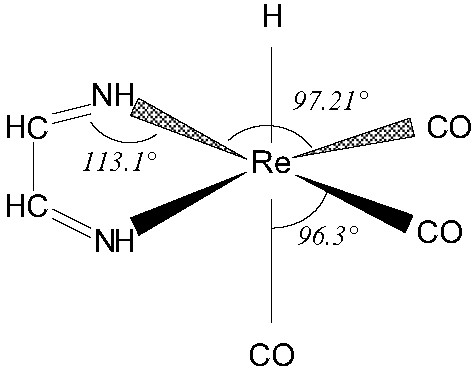
Idealized geometry of HRe(CO)3(H-dab) [5].
The details of the quantum chemical calculations are reported elsewhere [5].
The molecule has been modeled as a pseudo diatomic with the hydrogen on one side and the rest of the complex on the other side. The Re–H bond distance q is the only degree of freedom; all other coordinates were remained fixed and were decoupled from q.
The photo-absorption and emission dynamics were obtained by solving the time-dependent Schrödinger equation,(1)
The kinetic part of the Hamiltonian of the system is given by:(4)
The absorption and emission spectra are obtained by evaluation of the cross sections within the weak field approximation [8,9](5a)
The time-dependent emission spectrum is simulated by the 1A′ (GS) → b1MLCT absorption (see above), the wave packet developed on the absorbing state being transferred on the electronic ground state at each step of the propagation on the upper state.
3 Absorption/emission spectra of HRe(CO)3(H-dab)
The CASSCF/MRCI potential energy curves calculated as a function of the Re–H bond elongation in HRe(CO)3(H-dab) [5] for the electronic ground state, the low-lying MLCT states and the 3SBLCT dissociative state are represented in Fig. 2. A detailed study of the theoretical absorption spectrum based on wave packet propagations on two-dimensional potential energy surfaces (PES) taking into account several low-lying singlet electronic states has been published elsewhere [5].
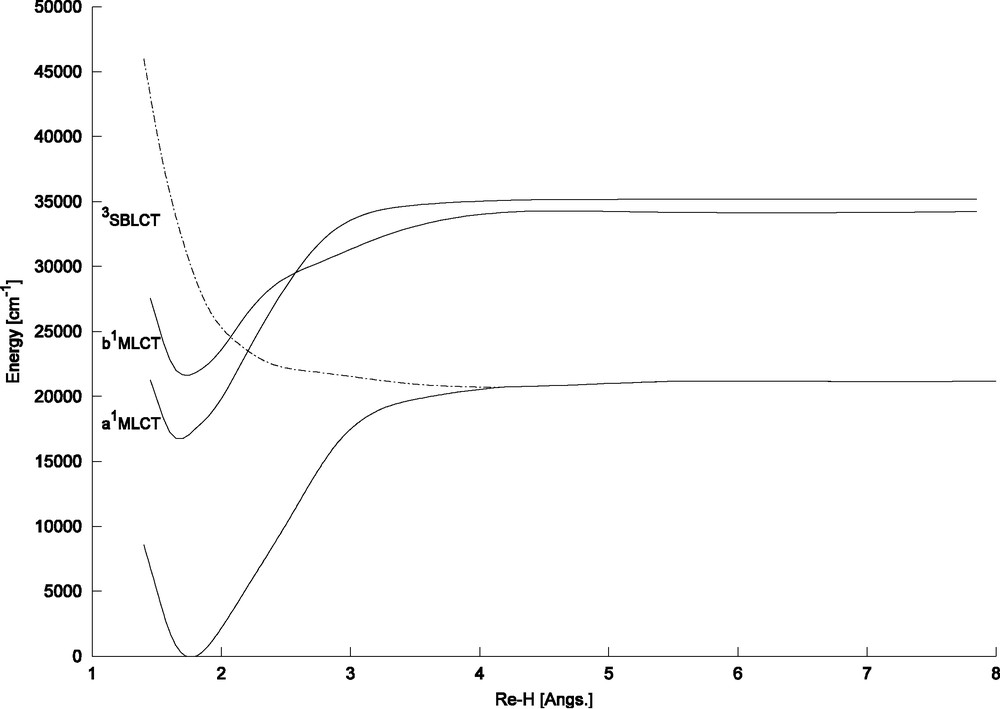
CASSCF/MRCI potential energy curves of HRe (CO)3(H-dab) calculated as a function of the Re–H bond elongation under Cs symmetry constraint [5] corresponding to the low-lying a1MLCT, b1MLCT (absorbing state) and 3SBLCT.
The goal of the present simulation based on a simple one-dimensional scheme including only the low-lying b1MLCT (the only one with a significant oscillator strength of 0.38) is to recover the main features of this absorption spectrum and to reproduce the characteristics of the corresponding emission spectrum originated from the absorbing state. The simulation should serve as a reference for the limiting case of a pure radiative decay. For this purpose the electronic ground state potential has been shifted by 0.4 a.u. to the short Re–H bond distances and the initial wave packet (Eq. (2)) prepared on the electronic ground state has been propagated on the upper MLCT state. The theoretical absorption spectrum is depicted in Fig. 3.
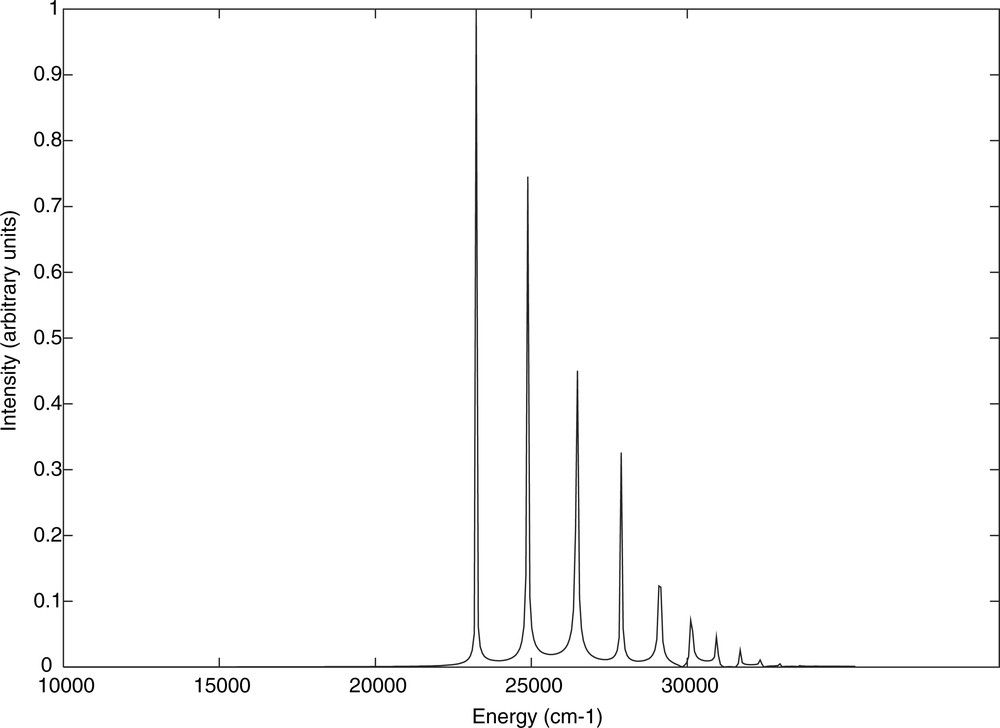
Theoretical absorption spectrum of HRe (CO)3(H-dab) corresponding to the electronic ground state → b1MLCT transition.
The simulated spectrum exhibits an intense peak at 23 200 cm–1 in agreement both with the experimental spectrum recorded for this class of molecules [10] and the published theoretical spectrum [5]. The shoulder at 35 000 cm–1 and the weak absorption at 15 000 cm–1 obtained in our previous simulation [5] do not appear in Fig. 3 since the corresponding low-lying singlet states are not included in the simulation. In order to compare the fine structure of the absorption and emission spectra we did apply a multiplication factor exp (–Γt, Γ = 0.10) necessary to get the envelope of the spectrum.
The induced emission spectrum built on the basis of the propagation of initial wave packets prepared on the absorbing state and defined by Eq. (3) is represented in Fig. 4. This emission signal reflects the structure of the absorption spectrum and is shifted to the red. The intense peaks between 20 000 and 25 000 cm–1 originate from the contribution of the initial Φ (q, Ek, n = 0) vibrational wavefunction corresponding to the vibrational ground state of the absorbing state (|0 > → |0 > at 23 200 cm–1). The initial Φ (q, Ek, n = 1) and Φ (q, Ek, n = 2) wave packets corresponding to excited vibrational levels lead to minor contributions of 0.5% and 0.01%, respectively, between 15 000 and 22 000 cm–1.
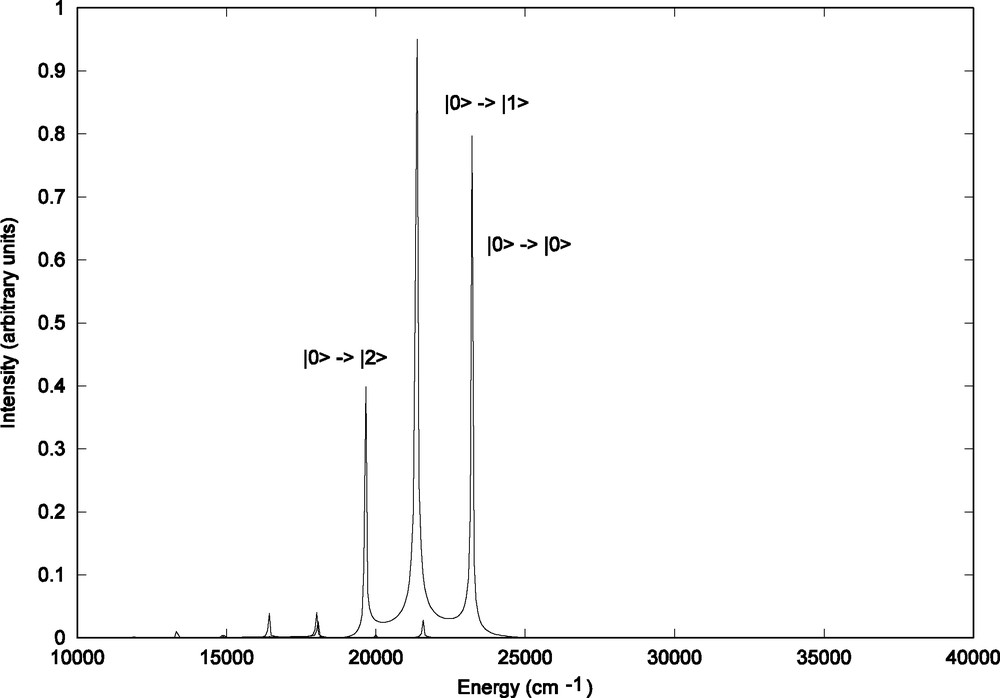
Total theoretical emission spectrum of HRe (CO)3(H-dab) corresponding to the b1MLCT → electronic ground state transition.
The structures of the partial contributions to the total emission spectrum are depicted in Fig. 5a–c. The contribution of the emission from the vibrational ground state (n = 0) of the b1MLCT electronic state (Fig. 5a) can be superposed to the total emission spectrum (Fig. 4) with three peaks corresponding to the and transitions.
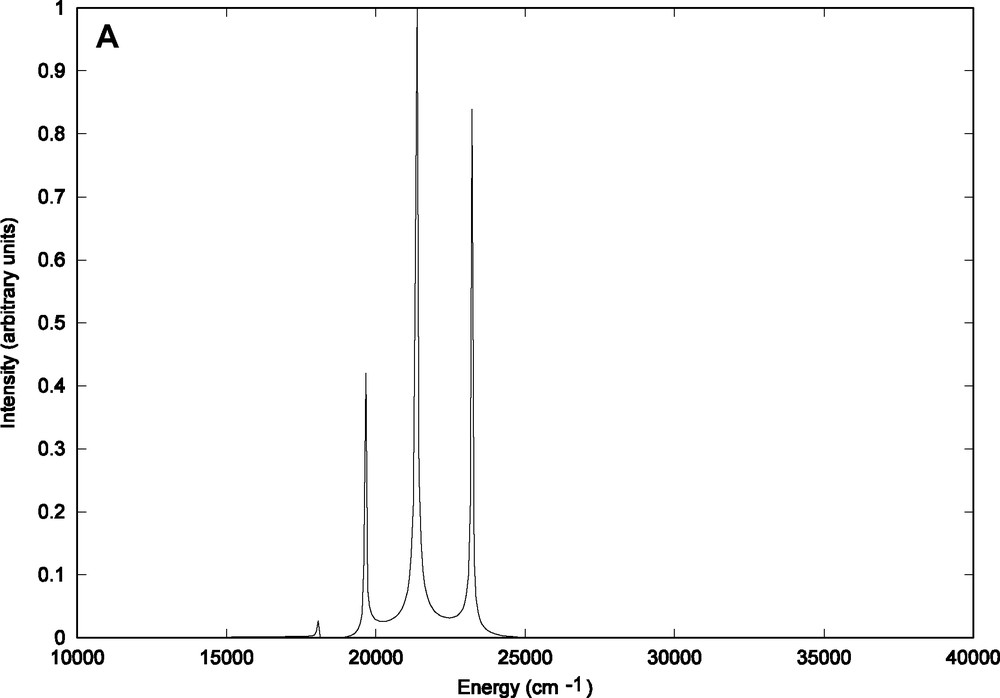

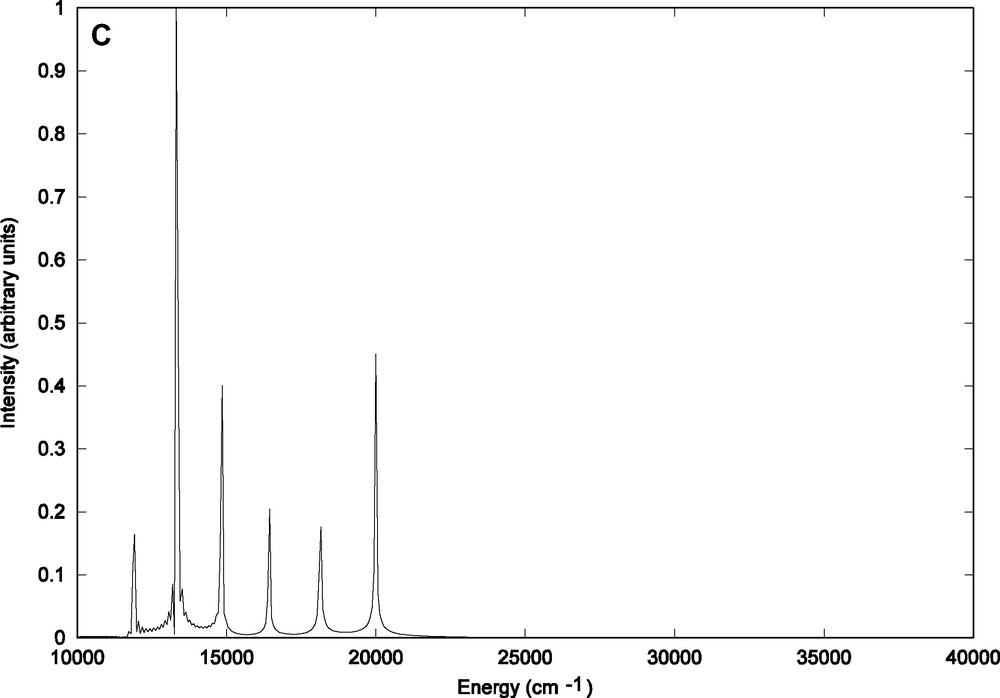
Contributions to the theoretical emission spectrum of HRe (CO)3(H-dab) from (A) the vibrational ground state (n = 0) of the b1MLCT state (B) the vibrational state (n = 1) (C) the vibrational state (n = 2).
Indeed this contribution does account for nearly 98% in contrast to the minor contributions reported in Fig. 5b (n = 1) and Fig. 5c (n = 2) which reveal very weak fine structures coming from transitions occurring from the initial Φ (q, Ek, n = 1) and Φ (q, Ek, n = 2) wave packets propagated on the upper potential (b1MLCT) to the various vibrational bound states of the electronic ground state.
4 Time-resolved emission spectrum of HRe(CO)3(H-dab)
The time-dependent emission spectrum simulated for the b1MLCT → electronic ground state transition in HRe(CO)3(H-dab) within a three-dimensional picture is represented in Fig. 6. In our simulation the laser promotes the initial state Φ (q, Ek, n = 0) multiplied by the corresponding transition dipole moment instantaneously to the upper b1MLCT absorbing state.
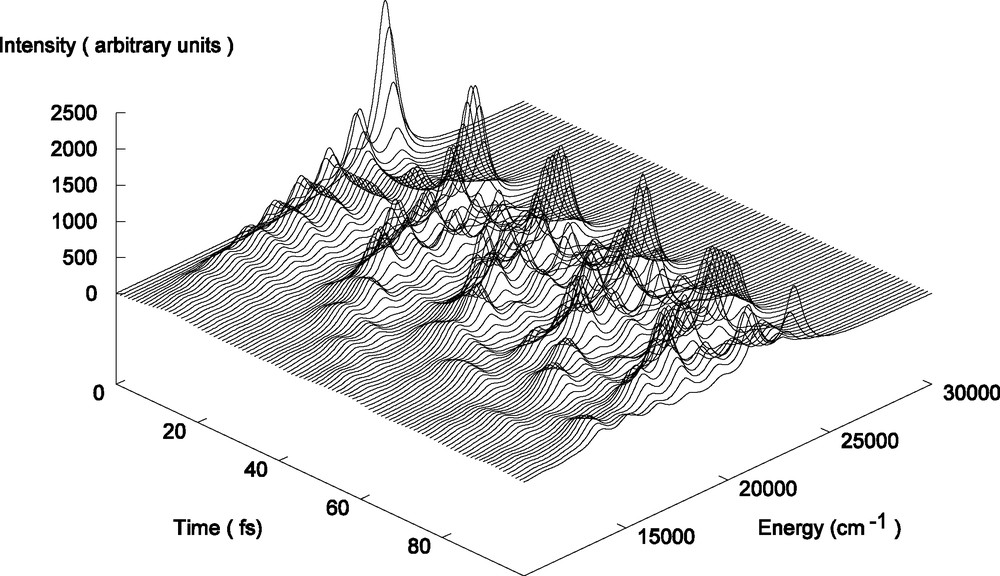
Time-dependent emission spectrum of HRe(CO)3(H-dab) simulated for the b1MLCT → electronic ground-state transition.
Under the influence of the Hamiltonian of the corresponding potential it starts to move and to accumulate overlap with all excited vibrational wavefunctions of the electronic ground state. The shape of the time-dependent emission spectrum points to the presence of an intense single peak at 23 210 cm–1 at the initial time which illustrates the Franck–Condon principle, namely the instantaneous nature of the electronic transition back to the ground state. In the few tens of femtoseconds the oscillatory motion of the wave packet in the potential well of the absorbing state leads to the formation of new lines to the lower energies corresponding to the population of the low-lying vibrational states of the emitting state. Within the same time scale a decrease in intensity of the initial peak is also observed which corresponds to the decay of the autocorrelation function S(t) as a function of time. After 40 fs the complicated pattern of the spectrum due to the ‘interference’ of the dynamics in the upper b1MLCT state and electronic ground state is difficult to analyze.
5 Conclusion
The absorption/emission spectra of [HRe(CO)3(H-dab)], model system for α-diimine rhenium carbonyls complexes, have been simulated by wave packet propagations on the one-dimensional potentials V(q = [Re–H]) calculated for the electronic ground state and the b1MLCT absorbing state. The main features of the absorption/emission spectra recorded for this class of molecules, namely an intense absorption peak in the visible and a corresponding emission band shifted to the red have been recovered. The emission spectroscopy has been simulated within the limiting case of a pure radiative decay, the other competing processes such as intersystem crossing, dissociation or vibrational relaxation in the medium being neglected. The time-resolved emission spectrum has been obtained by simulating the whole absorption/emission process on the basis of the decomposition of wave packet into a superposition of vibrational eigenstates v′, each with different phases. A sufficiently long time of propagation has been chosen in order to ensure the validity of the autocorrelation function Fourier Transform approach. In order to study the competition between the 1MLCT emission and the rhenium–hydrogen bond homolysis in [HRe(CO)3(H-dab)] it will be necessary to go beyond the present theoretical model based on the generalization of the Kramers–Heisenberg expression to the continuous spectrum [11].
Acknowledgements
The authors are grateful to Professor A. Strich and Professor J. Manz for helpful discussions. The simulations have been carried out at the IDRIS (Orsay, France) through a grant of computer time from the Conseil Scientifique.