

1 Introduction
Processes leading to linear α-olefins have gained much commercial significance owing to their various applications, in particular in the fields of plasticizers and detergents [1,2]. New grades of linear low-density polyethylene (LLDPE) have been developed, which are obtained by copolymerization of ethylene with a light α-olefin, such as 1-butene, 1-hexene or 1-octene. Current processes for the synthesis of linear α-olefins are based on the oligomerisation of ethylene and the ability to modulate the carbon-chain length remains a central issue [3]. Typical distributions cover the C4–C28 (Ethyl Corporation) and C4–C50 ranges (Chevron, Shell). Although modulation of chain length can be achieved by a subsequent olefin metathesis reaction, it remains important to develop catalysts able to oligomerize ethylene selectively in α-olefins in the C4–C8 range.
Nickel(II) complexes containing specific phosphorus–oxygen chelating ligands are very efficient catalysts for the oligomerisation of ethylene to linear α-olefins [4–8]. The combined soft and hard donor character of the chelating P,O ligand plays a key role in determining the properties of these catalysts that have led to the industrial Shell Higher Olefin Process (SHOP) for the selective production of liquid linear α-olefins, which is one of the largest applications of homogeneous catalysis [8]. The structurally characterized complex (1a) [9], which contains both a Ni-carbon σ-bond and a bidentate, three-electron-donor P∩O chelating ligand, catalyzes the oligomerisation of ethylene at 50 bar and 50 °C, and is regarded as a suitable model for the SHOP catalyst [7,8].

Such square planar Ni(II) complexes are typically prepared by oxidative addition of (benzoylmethylene)triphenylphosphoranes Ph3P=CHC(=O)R to Ni(0) complexes such as [Ni(cod)2] (cod = 1,5-cyclooctadiene), in the presence of a two-electron-donor phosphine ligand. It is remarkable that the oxidative addition of a P-phenyl bond across the zerovalent metal centre has so far remained limited to nickel. To access Pd(II) complexes analogous of the Ni-based SHOP catalysts, we therefore had to develop another approach, which consisted in the coordination of a functional phosphine of the type Ph2PCH2C(=O)R to a Pd(II) aryl complex, followed by deprotonation of the coordinated phosphine to generate the phosphinoenolate complex [10]. Since subtle variations of the ligands may be sufficient to change significantly the catalytic properties of these complexes, we were interested in modifying the stereoelectronic environment of the nickel centre. We could show that the α-iminophosphorus ylide Ph3P=CHC(=NPh)Ph 2 also oxidatively adds to [Ni(cod)2] and affords P,N analogues of the P,O systems. Depending upon the nature of the added two-electron-donor phosphine ligand, coordination of the latter or of an intact ylide molecule was observed, as in 3 or 4, respectively (Scheme 1) [11].
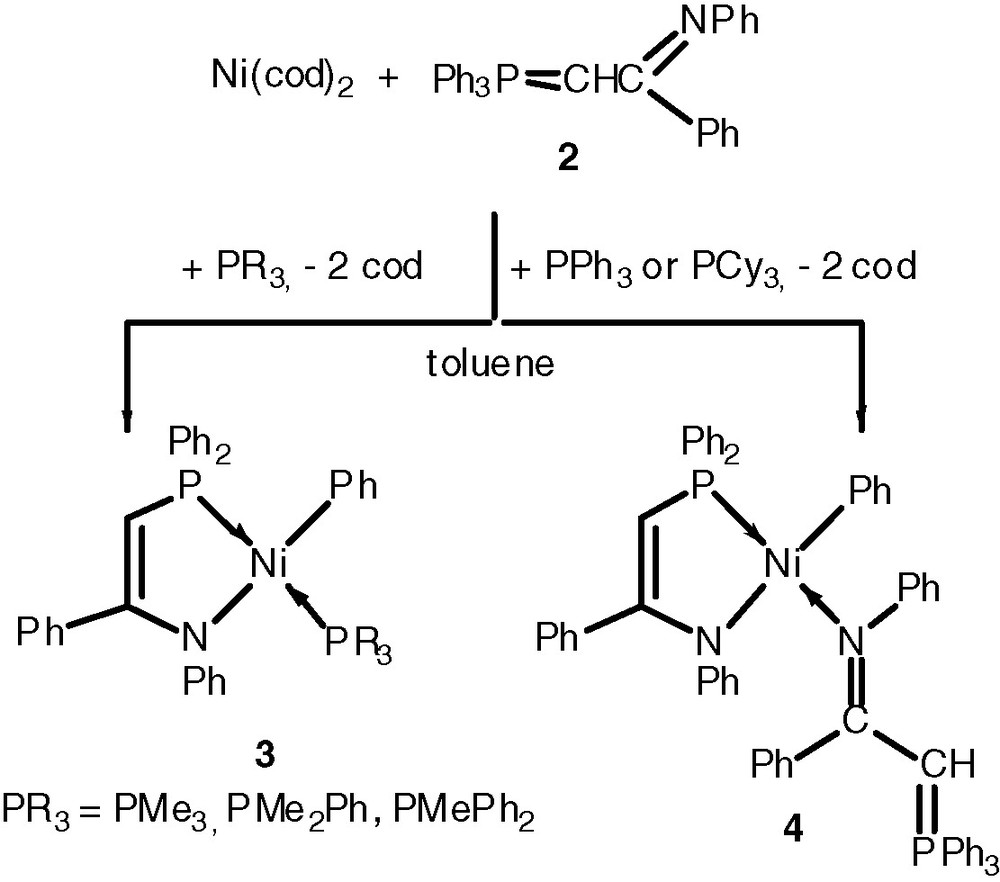
Similar observations were made with isoelectronic azaphosphoranes (Scheme 2) [11b].
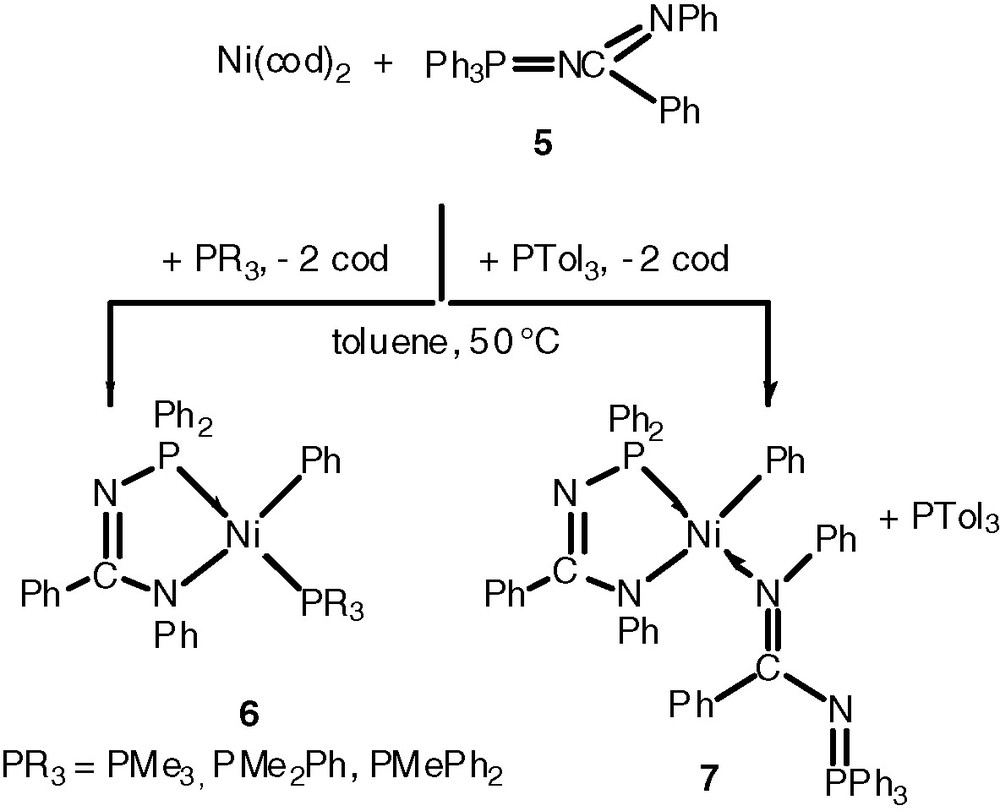
The presence of a nitrogen donor atom in 3, 6 or (8) instead of oxygen in the three electron donor chelate was found to be detrimental for the catalytic activity [7,11a].
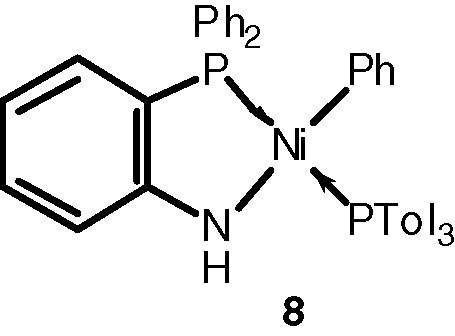
However, recent results have shown that Ni(II) complexes containing neutral, four-electron-donor P,N chelates (P = phosphine, phosphonite or phosphinite moiety; N = pyridine or oxazoline donor) are efficient and selective dimerisation or oligomerisation catalysts in the presence of MAO or small amounts of AlEtCl2 as cocatalyst [12].
The activity and selectivity (α-olefin distribution) of complexes of type 1 are strongly dependent on the electronic and steric properties of the substituents, the ring size of the chelate and the nature of the two-electron-donor ligand. As shown in Scheme 3, dissociation of the phosphine ligand enables coordination of an ethylene molecule that can then undergo insertion into the Ni–Ph (initiation step), Ni–H or Ni-alkyl bond and this leads to chain growth.
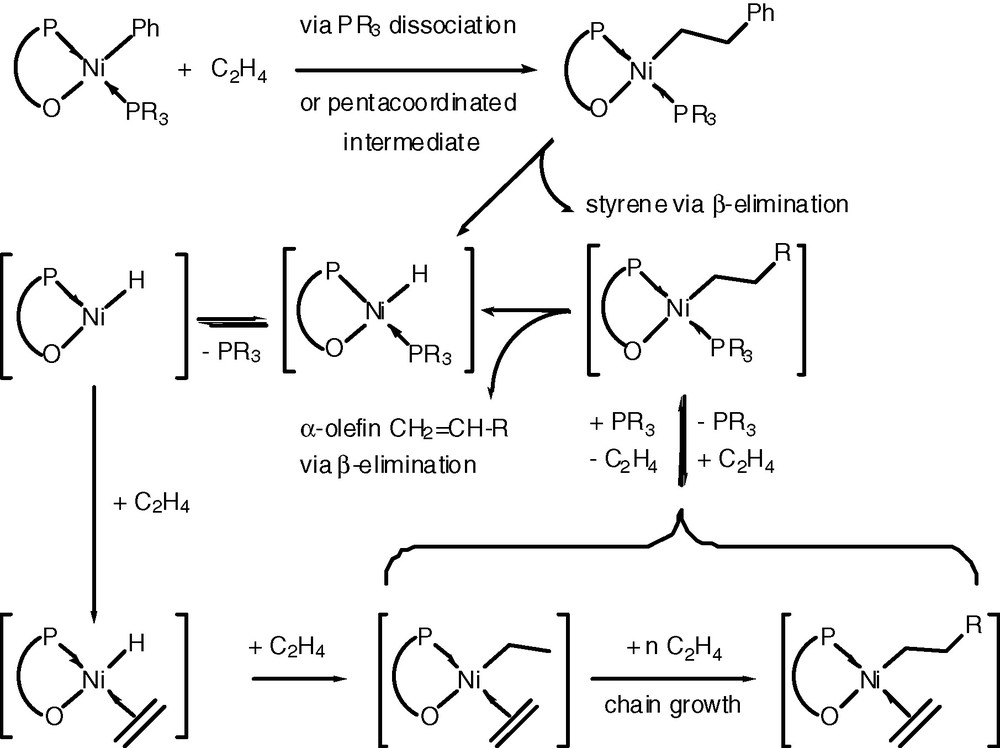
There have been numerous attempts to modify the molecular weight distribution of the resulting oligomers by changing the structure of the phosphinoenolate ligand and/or the ancillary phosphine [8a,13]. With the aim of facilitating β-hydrogen elimination during the growing step (Scheme 3) and thus favour low-molecular-weight products, we have attempted to modify the electronic density at the nickel centre by favouring the occurrence of an additional intramolecular bonding interaction between the enolate oxygen atom and an ortho-substituent on the aromatic ring of the P,O chelate, as shown with 9–11 [5].
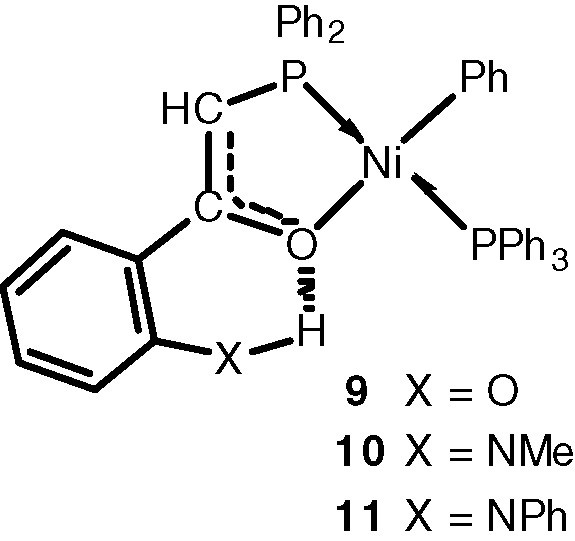
Here we report further studies on the influence of the substituents at X = N and of the phosphine PR3 on the structural and catalytic properties of the corresponding Ni(II) complexes. We also compare the crystal structures of the new complex 16, which contains a P(p-C6H4F)3 ligand, with those of the PPh3 complexes 10 and 11.
2 Results and discussion
Reaction of the (ortho-substituted benzoylmethylene)triphenylphosphoranes 12a–c, prepared in a manner similar to related ylides [14], with one equivalent each of [Ni(cod)2] and triarylphosphine in toluene, afforded the respective Ni(II) complexes under similar conditions to those described previously for complex 11 (Scheme 4) [5,9]. The new yellow complexes were isolated analytically pure by precipitation with pentane. The trans arrangement of the P atoms gives rise in the 31P{1H} NMR spectrum to an AB pattern with typical coupling constants.
A view of the crystal structures of complexes 10 and 16 is shown in Figs. 1 and 2, respectively, and selected bond distances and angles are given in Table 1. The square-planar coordination around the Ni(II) centre is only slightly distorted and the five-membered Ni–P–C–C–O cycle is nearly planar, owing to electron delocalisation within the enolate moiety. The relatively short Ni–Ph distance is consistent with the electron affinity of the P,O chelating ligand. Since the first step in the catalytic cycle is the insertion of ethylene into the Ni-phenyl bond, which is followed by β-H elimination, the strength of this bond influences the catalyst’s activation temperature [8a]. The most notable feature in these structures is the strong intramolecular N–H···O hydrogen bonding: the calculated distance between the NH hydrogen atom and the oxygen atom of the enolate ligand is rather short: 2.18(5) Å in 10·2 (C7H8) and 2.00(5) Å in 16·1/2 (C6H6), respectively [15,16].

Molecular structure of complex 10 in 10·2 (C7H8). Except for N–H(1), the H atoms are not represented.
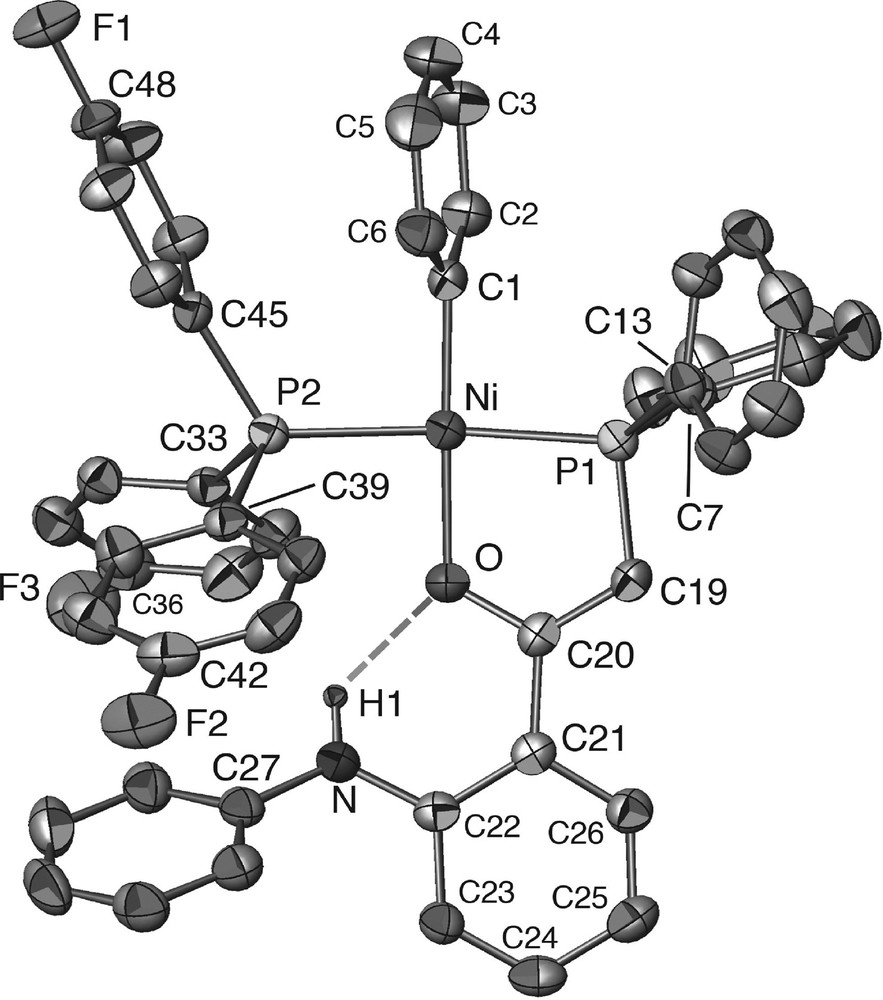
Molecular structure of complex 16 in 16·1/2 (C6H6). Except for N–H(1), the H atoms are not represented.
Selected bond lengths (Å) and angles (deg) in complexes 10·2 (C7H8) and 16·1/2 (C6H6), compared with those for 11 [5]
| 10·2 (C7H8) | 16·1/2 (C6H6) | 11 | |
| Ni–C(1) | 1.899(6) | 1.893(5) | 1.886(3) |
| Ni–P(1) | 2.183(2) | 2.161(2) | 2.180(1) |
| Ni–P(2) | 2.223(2) | 2.213(2) | 2.214(1) |
| Ni–O | 1.908(4) | 1.929(4) | 1.923(2) |
| P(1)–C(19) | 1.772(6) | 1.762(5) | 1.766(4) |
| C(19)–C(20) | 1.354(8) | 1.360(8) | 1.345(4) |
| C(20)–C(21) | 1.497(8) | 1.489(7) | 1.498(5) |
| C(21)–C(22) | 1.410(8) | 1.415(8) | 1.413(4) |
| C(22)–N | 1.371(8) | 1.381(7) | 1.396(5) |
| N–C(27) | 1.441(9) | 1.409(7) | 1.400(4) |
| C(20)–O | 1.315(7) | 1.327(6) | 1.332(4) |
| P(1)–Ni–P(2) | 174.60(7) | 172.43(6) | 159.78(4) |
| C(1)–Ni–O | 175.7(2) | 178.5(2) | 170.1(1) |
| O–Ni–P(1) | 85.8(1) | 86.3(1) | 86.20(6) |
| P(1)–C(19)–C(20) | 113.3(4) | 114.7(4) | 113.6(2) |
| C(19)–C(20)–C(21) | 122.9(5) | 123.1(5) | 123.1(3) |
| C(19)–C(20)–O | 121.7(5) | 120.3(5) | 122.4(3) |
| C(20)–O–Ni | 119.8(3) | 119.0(3) | 117.9(2) |
| C(21)–C(22)–N | 120.8(5) | 121.8(5) | 119.8(3) |
| C(22)–N–C(27) | 122.2(6) | 125.4(5) | 128.3(3) |
The coordination geometry around the nitrogen atom in complexes 10·2 (C7H8) and 16·1/2 (C6H6) is planar, with the sum of their valency angles being 360.0(5)° and 359.7(5)°, respectively. This was also observed in the structure of 11 and is probably due to the combined influence of electronic and steric interactions resulting from the hydrogen bonding which leads to a stabilised, six-membered ring. The 1H NMR data are typical for NH···O protons [5]. To evaluate the consequences of changing one parameter at a time in the ligand system on the main bonding parameters, a comparison of the structural data for complexes 10·2 (C7H8), 11 and 16·1/2 (C6H6) is provided in Table 1. Only minor variations are observed when the nitrogen substituent is Me or Ph (compare 10 and 11) or when the PPh3 ligand is replaced with the less electron donor P(p-C6H4F)3 (compare 11 and 16). However, we will see that the effects are more pronounced when looking at the catalytic properties of these complexes.
The distribution of oligomers obtained with the various catalyst precursors is shown in Fig. 3 and selected catalytic data are presented in Table 2. Complexes 9–11 and 13–16 display high activity and selectivity in ethylene oligomerisation to linear α-olefins (20 ml toluene, 0.10 mmol catalyst, 60–80 °C, 55 bar ethylene, 1.5–6 h), with a marked shift of the product distribution to lower molecular weights when compared to 1a under similar experimental conditions. A comparison between the complexes containing the PPh3 ligand shows that the effect of hydrogen bonding on product distribution is more pronounced for 9 and 11 than for 10 and 13. Comparing pre-catalysts 11, 15 and 16 shows that the influence of the phosphine ligand on the mass distribution appears modest, except in the C4 fraction whose selectivity increases to 37% in the case of the P(p-C6H4F)3 derivative 16, in contrast to its larger influence on the productivity and TOF values (see Table 2). Complex 15 yielded a broader mass distribution and heavier olefins were obtained (waxes).
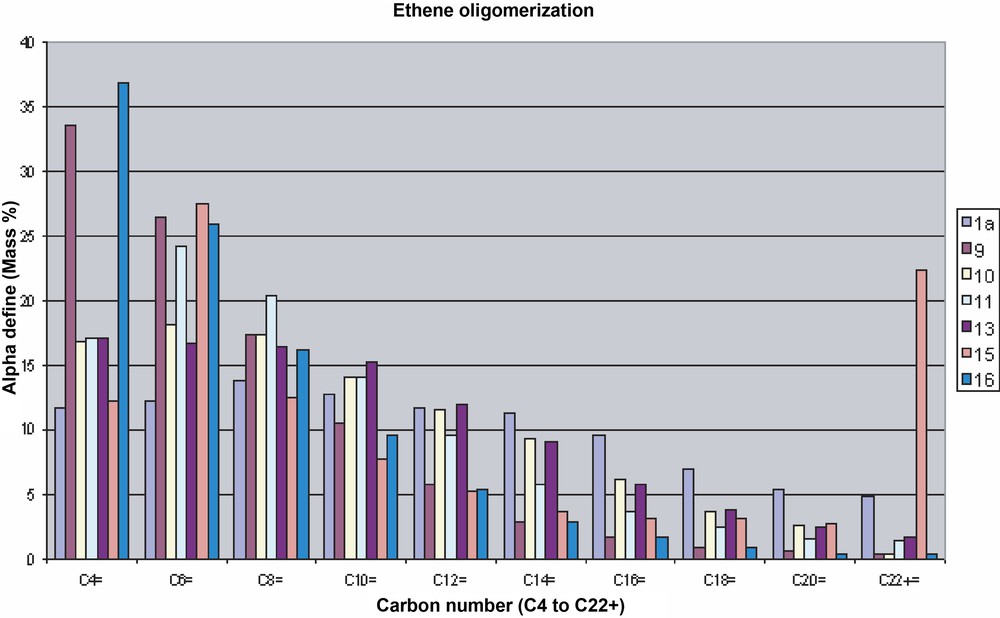
Selectivity in mass% of the oligomers formed in the catalytic reactions. For each carbon fraction, the sequence of complexes is: 1a, 9, 10, 11, 13, 15 and 16.
Catalytic data of the nickel complexes in the oligomerisation of ethylene
| Complex | T (°C) | Time (h) | kα (SF)a | Productivityb | TOFc | Selectivity (%)d |
| 1a | 60 | 1.5 | 0.7 | 2.7 | 5700 | 98 |
| 9 | 60 | 1.5 | 0.48 | 2.7 | 5700 | 89–92 |
| 10 | 70 | 4 | 0.62 | 1.3 | 2700 | 96–98 |
| 11 | 80 | 3.5 | 0.56 | 2.9 | 6100 | 97–99 |
| 13 | 60 | 5.5 | 0.61 | 2.3 | 4800 | 98–99 |
| 14 | 80 | 6 | 0.88 | 5.3 | 11 100 | nde |
| 15 | 70 | 4 | 0.82 | 6.3 | 13 200 | nde |
| 16 | 67 | 1.5 | 0.47 | 4.2 | 8800 | 97–98 |
a The kα factor is calculated without considering the butenes.
b kg of C2H4 (g Ni h)–1.
c mol of C2H4 (mol Ni h)–1.
d Selectivity for linear α-olefins (C4 to C8).
e Partial loss of light olefins during the dissolution of the waxes.
We have previously observed that replacing PPh3 with PCy3 in complexes of type 1 leads to an increase of the TON from 82 000 to 180 000 mol C2H4 converted·(mol catalyst h)–1 [7]. A similar trend is observed here when comparing the data between 13 and 14 or 11 and 15.
The high selectivity observed for linear α−olefins is consistent with the ethylene pressure used, which provides a high-enough concentration in solution to limit efficiently consecutive, codimerisation reactions. The complex analogous to 9 with a methoxy group in ortho position instead of a OH group yielded a productivity of 2.2 kg C2H4 (g Ni h)–1 and a TOF of 4600 mol C2H4 (mol Ni h)–1, slightly lower than 9, but gave a maximum mass distribution for the C8 and C10 fractions, similar to that observed with 1a, suggesting no significant electronic influence of the MeO substituent. Obviously, the intramolecular hydrogen bonding in these new catalytic precursors allows a fine tuning of their selectivity; it favours β-hydrogen elimination and thus the formation of C4–C8 olefins, of which 95–99% are linear α-olefins. Further work is still needed to fully evaluate and rationalize the catalytic implications of such intramolecular ligand effects.
3 Experimental section
3.1 Reagents and physical measurements
All operations were performed in Schlenk-type flasks under high-purity argon, using vacuum line techniques. The solvents were purified and dried under argon by conventional methods. The 1H NMR spectra were recorded at 200 MHz on a Bruker AC 200 F, the 31P{1H} NMR spectra at 81 MHz on a Bruker CXP 200 and at 122 MHz on a Bruker AC300 instrument. The spectra were recorded at room temperature and the 1H and 31P shifts are given relative to internal TMS and external H3PO4, respectively. A positive sign denotes a shift downfield from that of the reference. Coupling constants are given in Hz. The electron impact mass spectra (EI, 70 eV) were recorded on a Fisons ZAB-HF spectrometer. Reactions with ethylene were performed in a 130-ml double-walled stainless steel autoclave, fitted with a manometer, a septum inlet and a magnetic stirrer. The products were analysed by gas phase chromatography with a Hewlett Packard 5890 Series II instrument on a PONA column (methylsilicone, diameter: 0.22 mm, length: 50 m, temperature program from 35–270 °C) for the C4=–C30= fraction, and on a capillary Supelco Polywax SPB1 column (diameter: 0.53 mm, length: 30 m, ‘on-column injection’, 3 min at 5 °C, then temperature program 10 °C min–1 up to 380 °C) for the C4=–C100= fraction. The highest mass detected corresponded to C100=. High-purity ethylene was purchased from Air Liquide and used as received.
3.2 Synthesis
The complexes [Ni(cod)2] [17], 10 and 11 [5] were synthesized according to the literature and complexes 13–16 were prepared according to Scheme 4 by following the method used for 10 and 11. Satisfactory elemental analyses were obtained for the new complexes. The phosphine P(p-C6H4F)3 was purchased from Strem and used as received.
Selected data for 10: IR (KBr): 1600w, 1580w, 1510s, 1470w cm–1. 1H NMR (200 MHz, C6D6) δ: 1.90 (s, 3H, NMe), 5.05 (s, 1H, PCH). 31P{1H} NMR (81 MHz, C6D6): 16.4 (d, 2J(PP) = 280 Hz, PPh3), 19.5 (d, P,O).
13: R (KBr): 1600w, 1555s, 1475w cm–1. 1H NMR (200 MHz, C6D6) δ: 5.60 (2H, br s, NH2), 6.10 (s, 1H, PCH). 31P{1H} NMR (81 MHz, C6D6): 15.6 (d, 2J(PP) = 281 Hz, PPh3), 20.4 (d, P,O).
14: 1H NMR (200 MHz, C6D6) δ: 5.60 (2H, br s, NH2), 6.10 (s, 1H, PCH). 31P{1H} NMR (81 MHz, C6D6): 13.9 (d, 2J(PP) = 265 Hz, PCy3), 17.9 (d, P,O).
15: IR (KBr): 1585w, 1565, 1510s, 1495w, 1455w cm–1. 1H NMR (200 MHz, C6D6) δ: 5.60 (2H, br s, NH2), 6.10 (s, 1H, PCH). 31P{1H} NMR (81 MHz, C6D6): 13 (d, 2J(PP) = 266 Hz, PCy3), 17.4 (d, P,O).
16: IR (KBr): 1590w, 1570w, 1495s, 1455w cm–1. 1H NMR (200 MHz, C6D6) δ: 5.10 (s, 1H, PCH), 6.35–7.75 (m, 36H, aromatic), 8.80 (s, 1H, NHPh). 31P{1H} NMR (81 MHz, C6D6): 16 (d, 2J(PP) = 284 Hz, P(p-C6H4F)3), 17.2 (d, P,O).
3.3 Crystallography
Single crystals suitable for X-ray diffraction were obtained by recrystallisation of 10 and 16 from toluene/hexane or benzene/hexane, which afforded 10·2 (C7H8) and 16·1/2 (C6H6), respectively. Diffraction data were collected on a Philips PW1100/16 diffractometer the using Cu Kα graphite monochromated radiation (λ = 1.54184 Å) by θ/2θ flying step-scan. The structures were solved using SHELXS-97 and full-matrix least-square refinements were carried out using the SHELXL-97 program [18]. All non-hydrogen atoms were refined with anisotropic parameters. The hydrogen atoms were included in their calculated positions and refined with a riding model. The benzene molecule found in 16·1/2 (C6H6) was disordered. For both structures, atomic coordinates, bond lengths and angles, and thermal parameters have been deposited within the Cambridge Crystallographic Data Centre, CCDC 248694 and 248970.
3.3.1 Crystal data for compound 10·2 (C7H8)
Yellow crystals from toluene/hexane. C45H39NNiOP2·2 C7H8, M = 914.8; monoclinic; space group P21/n (No. 14); a = 20.9190(10), b = 14.7780(10), c = 15.7050(10) Å, β = 97.22(5), V = 4816.6(7) Å3, Z = 4, Dc = 1.261 g cm−3, μ = 1.523 mm–1, F(000) = 1928, T = 173 K; 5058 unique reflections (θ = 3.3–51.1 °), of which 4359 with I > 2 σ(I) were used for structure solution; R = 0.0726, wR2 = 0.2061.
3.3.2 Crystal data for compound 16·1/2 (C6H6)
Yellow crystals from benzene/hexane. C50H38F3NNiOP2·1/2C6H6, M = 885.52; triclinic; space group P (No. 2); a = 12.248(2), b = 17.882(2), c = 10.365(2) Å, α = 101.67(5), β = 103.12(5), γ = 80.73(5), V = 2149.3(8) Å3, Z = 2, Dc = 1.368 g cm–3, μ = 1.794 mm–1, F(000) = 918, T = 173 K; 4329 unique reflections (θ = 2.5–50.0°), of which 3981 with I > 2 σ(I) were used for structure solution; R = 0.0633, wR2 = 0.1880.
Acknowledgements
Financial support from the ‘Centre national de la recherche scientifique’ and the ‘Institut français du pétrole’ (IFP) is gratefully acknowledged. We are grateful to Drs D. Commereuc, H. Olivier (IFP) for numerous discussions and to Dr. A. DeCian and Prof. R. Welter (‘Service de rayons X’, Strasbourg) for the determination of the crystal structures.