Owing to their ability to form rather stable [1] inclusion complexes with hydrophobic guest, on the one hand, and to their solubility both in water [2] and organic solvents [3], on the other hand, per-O-methylated β-cyclodextrins (CDs) are widely used in several areas of science and technology [4]. The controlled chemical synthesis of methylated CDs having a few selectively located non-methylated positions ready for further chemical modifications is thus highly desirable. We have recently described a straightforward solution to this problem, that is the treatment in toluene of commercially available permethylated β-cyclodextrins with diisobutylaluminum hydride (DIBAL). In doing so, we first observed a regioselective bis-de-O-methylation on the secondary rim, giving the 2A, 3B diol in 55% yield [5]. In a further step, increasing the amount of DIBAL and kinetically controlling the reaction, a 2A, 3B, 2D, 3E tetrol could easily be obtained in 51% yield [6]. These processes are remarkable in several ways: firstly, the yields are quite satisfactory and overwhelmingly higher than the ones attainable through a classical multistep process [7]; secondly, the compounds are very easy to obtain in pure form through a regular silica-gel column chromatography. Finally, it is important to point out that the behaviour of polymethylated β-CD is strikingly different from the one of polybenzylated β-CD, wherein only the primary rim is selected for de-O-alkylation [8]. Furthermore, the precise mechanism of these sequential bis-de-O-methylation processes is not known, a feature contrasting with our rather clear picture of the de-O-benzylation reaction [9].
Now, using a longer reaction time, we discovered that a hexol could be directly obtained in a preparative yield. When permethylated β-cyclodextrin 1 was treated with 50 equiv of DIBAL in toluene at room temperature for 28 h, hexahydroxy heptakis (2, 3, 6-tri-O-methyl) β-cyclodextrin 2 was obtained in a satisfactory yield (45%) after an easy separation by silica-gel chromatography (Scheme 1)1. Caution should be taken to keep the reaction system rigorously anhydrous.
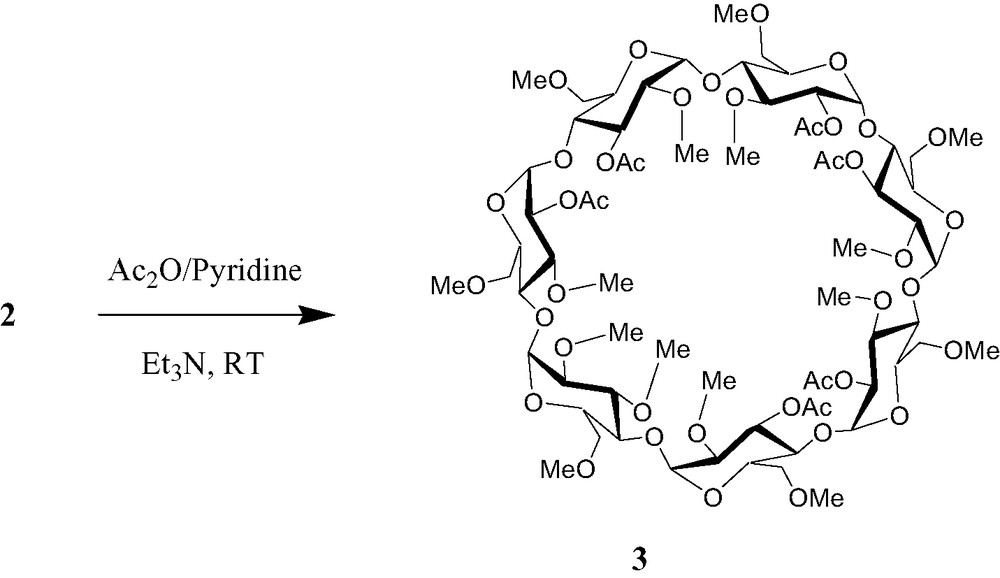
Further acetylation of 2 gave the hexaacetyl heptakis (2, 3, 6-tri-O-methyl) β-cyclodextrin 3 (Scheme 2)2. 1H NMR spectrum of 3 showed six acetyl groups at δ 2.14, 2.16, 2.17, 2.18, 2.19 and 2.20 ppm, while 13C NMR spectrum displayed six methyl and carbonyl groups of acetate at δ 21.10, 21.16, 21.17, 21.28, 21.29, 21.36 and 169.60, 169.70, 170.44, 170.50, 170.55 ppm, respectively. In addition, two sets of protons in 3, each referring to 3H, showed obvious downfield shifts as compared to 1H NMR of 2, appearing at 4.69–4.74 (3 H) and 5.39–5.46 (3H) ppm respectively, due to acetylation of the hydroxy groups. Further investigation on the 1H-1H and 1H-13C COSY exhibited that they should be assigned to three 2-O and three 3-O position protons.
The structural determination of 2 was achieved through the analysis of homonuclear 2D NMR spectra: COSY, RELAY, double-RELAY and off-resonance ROESY [10] (effective angle = 54.7°, mixing time 100 ms), as well as a HSQC heteronuclear spectrum. Six hydroxyl proton signals were observed and correlated to six protons by correlations on COSY spectra. Correlations to six different H-1 protons on RELAY and double-RELAY spectra assessed that the six hydroxyl substituents are carried by six different glucopyranose units and are positioned on carbon 2 for three of them (named A, C and E), and on carbon 3 for the three others (named B, D and F). Three OH2–OH3 correlations were observed on the off-resonance ROESY spectrum assessing the A→B, C→D and E→F links. The unique β-cyclodextrin derivative compatible with the previously mentioned NMR observations is the one described in Scheme 1 and in a simplified version in Fig. 1. Assignment of H1–H3 protons of the permethylated glucopyranose unit was straightforwardly performed by COSY, RELAY, and double-RELAY spectra. C-1 and C-2 were assigned on HSQC spectra (see Table 1).

Schematic version of hexahydroxy permethylated cyclodextrin 2. Red circles correspond to A, C and E units in an undetermined order, and consecutive clockwise blue circles correspond respectively to B, D and F units. The black circle corresponds to the permethylated unit G.
13C and 1H NMR (500 MHz) chemical shiftsa for 2 in DMSO-d6 at 25 °C
| A | B | C | D | E | F | G | |
| C1 | 105.56 | 103.16 | 105.53 | 102.91 | 105.75 | 103.00 | 102.19 |
| C2 | 76.88b | 85.20c | 76.88b | 85.20c | 76.88b | 85.20c | 85.57 |
| H1 | 5.380 | 5.606 | 5.385 | 5.611 | 5.390 | 5.615 | 5.619 |
| H2 | 3.941 | 3.611 | 3.951 | 3.610 | 3.946 | 3.607 | 3.653 |
| H3 | 3.817 | 4.301 | 3.844 | 4.309 | 3.806 | 4.301 | 3.882 |
| H4 | — | 3.978 | — | 3.985 | — | 3.966 | 4.018 |
| OH2 | 6.276 | — | 6.250 | — | 6.393 | — | — |
| OH3 | — | 6.546 | — | 6.477 | — | 6.442 | — |
a 1H and 13C methoxy signals are observed through HSQC spectra but remain not assigned.
b 1H–13C correlation peaks are not discernible.
c 1H–13C correlation peaks are not discernible.
In conclusion, we have been able to regioselectively eradicate six methyl groups from commercially available permethylated β-cyclodextrin in satisfactory yield, without any complicated purification steps. This hexahydroxy cyclodextrin is now adding to the dihydroxy [5,11] and tetrahydroxycyclodextrins [6] previously obtained by us, using the same molecular scalpel. Selecting the appropriate concentration of DIBAL and the duration of the reaction, one of these three products are now easily preparable in one step, and in about 50% yield. For the first time, a battery of new β-cyclodextrin derivatives is thus now accessible in a preparative and expeditious manner. Such a research will hopefully stimulate their use in technological applications.
Acknowledgements
Y. Chen thanks the French Ministry of Research and Technology for post-doctoral fellowship. We are grateful to Cyclolab (Hungary) for a generous supply of starting material 1, and Drs H. Desvaux and P. Berthault for technical assistance and valuable discussions.
1 Synthesis of hexahydroxyl-per-O-methyl-β-cyclodextrin (2). To a solution of heptakis (2,3,6-tri-O-methyl)-β-cyclodextrin (200 mg, 0.14 mmol) in anhydrous toluene (12 ml) was added 4.67 ml (7 mmol, 50 equiv) of DIBAL (1.5 M in toluene). The reaction mixture was stirred at room temperature for 28 h under argon. After quenching the reaction by adding an aqueous HCl (1 M), the toluene phase was separated, and the water phase was concentrated to 1/4 of the original volume. The residue was extracted by ethyl acetate (50 ml), the combined organic phase was washed by brine, dried with MgSO4, and evaporated to dryness. The resultant solid was subjected to silica gel chromatography eluting with CH2Cl2/MeOH (94:6, v/v) to give 85 mg (yield 45%) of 2. Rf = 0.14 (CH2Cl2/MeOH 91:9); [α]D +156.7 (c 1.0, CHCl3); FAB-MS m/z: 1367 (M+Na+); 1H NMR (400 MHz, CDCl3): δ 3.93–3.12 (m, 97H), 4.94–4.95, (m, 3 × H-1), 5.03 (d, J = 3.3 Hz, H-1), 5.06 (dd, J = 3.4 Hz, 2 × H-1), 5.09 (d, J = 3.4 Hz, H-1); 13C NMR (100 MHz, CDCl3): δ 57.94, 58.09, 58.18, 58.20, 58.75, 58.77, 58.79, 58.90, 60.95, 61.07, 61.64, 61.95 (15 × OCH3), 70.21, 70.28, 71.20, 71.45, 71.51, 71.55, 71.60 (7 × C-5), 70.64, 70.93, 71.10, 71.20 (7 × C-6), 73.03, 73.19, 73.23, 81.09, 81.13, 81.20, 81.28, 81.40, 81.45, 81.56, 81.84, 82.05, 82.30, 82.74 (7 × C-2, 7 × C-3, 7 × C-4), 99.38, 99.61, 99.68, 99.73, 101.94, 102.12, 102.20 (7 × C-1); Anal. calcd for C57H100O35·2H2O: C 49.56, H 7.59. Found: C 49.26, H 7.46.
2 Synthesis of hexaacetyl-per-O-methyl-β-cyclodextrin (3). To a solution of 2 (28 mg) in pyridine/Et3N (6 ml, 1:1) was added Ac2O (3 ml) dropwise. The reaction mixture was stirred at room temperature for 18 h under argon. The solvent was removed in vacuo, and the residue was dissolved in CH2Cl2. The organic phase was washed with brine, dried over MgSO4, and evaporated to dryness. The resultant solid was subjected to silica-gel-chromatography eluting with CH2Cl2/MeOH (97:3, v/v) to give 25 mg (yield 76%) of 3. Rf = 0.63 (CH2Cl2/MeOH 91:9); [α]D +130.1 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 2.14 (s, 3H, CH3CO), 2.16 (s, 3H, CH3CO), 2.17 (s, 3H, CH3CO), 2.18 (s, 3H, CH3CO), 2.19 (s, 3H, CH3CO), 2.20 (s, 3H, CH3CO), 3.16–4.01 (m, 91H), 4.69–4.74 (m, 3H, 3 × H-2), 5.08–5.12 (m, 4H, 4 × H-1), 5.20 (d, J = 3.6 Hz, H-1), 5.21 (d, J = 3.6 Hz, H-1), 5.29 (d, J = 3.5 Hz, H-1), 5.39–5.46 (m, 3H, 3 × H-3); 13C NMR (100 MHz, CDCl3): δ 21.10, 21.16, 21.17, 21.28, 21.29, 21.36 (6 × CH3CO), 58.11, 58.14, 58.27, 58.49, 58.76, 58.93, 58.96, 61.32, 61.44, 61.51, 61.68 (15 × OCH3), 70.70, 70.79, 70.81, 70.93, 70.96, 71.16, 71.28 (7 × C-5), 70.55, 70.73, 70.93, 71.02, 71.10, 71.18, 71.41 (7 × C-6), 76.13, 76.33, 76.53, 76.68, 77.00, 77.32, 78.57, 79.26, 79.29, 79.30, 79.38, 79.53, 79.62, 79.83, 80.23, 80.67, 81.28, 81.96 (7 × C-2, 7 × C-3, 7 × C-4), 96.89, 96.92, 96.94, 98.10, 98.23, 98.70, 99.29 (7 × C-1), 169.60, 169.69, 169.70, 170.44, 170.50, 170.55 (6 × C=O); anal. calcd for C69H112O41: C 51.87, H 7.07. Found: C 51.84, H 7.17.