

1 Introduction
Le tétrathiofulvalène (TTF, Fig. 1), composé stable qui peut céder réversiblement deux électrons, a permis le développement d'une chimie extraordinairement variée. On peut citer, entre autres, la synthèse de complexes organiques présentant une haute conductivité électrique [1,2], la préparation de matériaux supraconducteurs [3], l'élaboration d'édifices supramoléculaires [4], ainsi que la synthèse d'un grand nombre de molécules dérivées du TTF [5]. Des dispositifs fondés sur ces molécules sont également étudiés, par exemple des transistors à effet de champ [6,7]. La littérature associée à cette chimie est extrêmement vaste, ce qui en rend la consultation ardue. Cependant, une difficulté d'un autre ordre, assez peu citée, se présente en ce qui concerne l'applicabilité de ces systèmes moléculaires : la préparation reproductible de phases pures dans l'état solide, si possible avec de bons rendements, n'est presque jamais une tâche aisée. En particulier, lors de la préparation d'un complexe à transfert de charge AnDm où A est un π-accepteur et D un π-donneur comme le TTF, le contrôle de la stœchiométrie n/m, la détection d'un éventuel polymorphisme et, finalement, la caractérisation structurale du mode d'empilement des molécules dans le solide, sont des étapes essentielles avant toute étude des propriétés physiques du complexe. En effet, des stœchiométries inattendues peuvent être obtenues, par exemple A1D7 avec le polyoxométallate β-[Mo8O26]4– comme accepteur [8,9]. Quant au polymorphisme, il n'est pas exceptionnel, un cas bien étudié étant celui du complexe métallique TTF-[Pd(dmit)2]2 où H2dmit est la 4,5-dimercapto-1,3-dithiol-2-thione [10], pour lequel cinq polymorphes ont été identifiés [11].

Formules développées de TTF et TNF.
Dans ce cadre, nous étudions des complexes où le donneur est le TTF et l'accepteur un dérivé de la 9-fluorénone, système peu étudié jusqu'à présent [12–16]. Avec un dérivé 9-dicyanométhylène du fluorène, nous avons récemment mis en évidence [16] un dimorphisme pour le complexe AD, qui affecte le degré de transfert de charge (δ). Nous présentons maintenant un nouveau cas de dimorphisme, pour le complexe A2D3, où l'accepteur est la 2,4,7-trinitro-9-fluorénone (TNF, Fig. 1). Nous déterminons également, de manière relative, le degré de transfert de charge dans ces complexes, en nous fondant sur des données de spectroscopie vibrationnelle.
2 Partie expérimentale
2.1 Préparation des complexes
Les composants TNF (Matheson) et TTF (Sigma) sont utilisés tels quels et l'acétonitrile employé comme solvant est de qualité chromatographique (Merck). Des solutions équimolaires sont préparées (3,42 × 10–5 moles dans 1,52 mL) et chauffées jusqu'à dissolution complète des réactifs, puis mélangées à chaud. Ce mélange est gardé dans un erlenmeyer étanche durant 19 heures, puis est réduit lentement, jusqu'à évaporation complète du solvant (environ 6 jours). On recueille alors des cristaux café, en forme de plaquettes, avec un rendement proche de 100%. Ces cristaux correspondent à la phase α du complexe (TTF)3(TNF)2. Si l'on réalise la cristallisation dans un tube à essai que l'on agite périodiquement pour dissoudre les cristaux formés jusqu'à obtenir une solution saturée (environ 10 jours), qu'on laisse alors se réduire lentement (environ 10 jours), on obtient des cristaux café de forme irrégulière, avec un rendement proche de 100 %. Ces cristaux correspondent à la phase β du complexe (TTF)3(TNF)2. Les cristallisations sont réalisées a 25°C et à l'abri de la lumière, sous atmosphère non contrôlée.
2.2 Cristallographie de monocristal
Les phases α et β ont été étudiées par diffraction des rayons X sur monocristaux. Les intensités de diffraction ont été mesurées à 23°C avec un diffractomètre Bruker P4 utilisant la radiation Mo-Kα (λ = 0,71073 Å) équipé d'un détecteur ponctuel [17] et les structures ont été résolues et affinées avec les programmes inclus dans le système SHELX [18].
α-(TTF)3(TNF)2 : C22H11N3O7S6; 0,65 × 0,20 × 0,12 mm3; triclinique, P, a = 7,3491(10), b = 11,1748(13), c = 15,497(2) Å, α = 90,017(10)°, β = 100,520(11)°, γ = 93,074(10)°, V = 1249,4(3) Å3, Z = 2 ; 8075 réflexions mesurées jusqu'à 2θ = 50°, 4404 réflexions indépendantes (Rint = 0,035), corrigées de l'absorption avec 15 réflexions mesurées à différents angles ψ (facteurs de transmission de 0,843 à 0,930). R1 = 0,041 pour 2551 réflexions avec I > 2σ(I) et wR2 = 0,109 pour toutes les réflexions, 343 paramètres étant affinés. Les atomes d'hydrogène ont été placés en positions calculées. Le pic résiduel le plus élevé dans la dernière carte de Fourier différence est de 0,202 e/Å3.
β-(TTF)3(TNF)2 : C22H11N3O7S6 ; 0,6 × 0,4 × 0,2 mm3; triclinique, P, a = 10,7584(13), b = 10,8277(13), c = 11,1429(12) Å, α = 89,292(8)°, β = 81,282(9)°, γ = 75,619(9)°, V = 1242,4(3) Å3, Z = 2 ; 9078 réflexions mesurées jusqu'à 2θ = 55°, 5695 réflexions indépendantes (Rint = 0,023), corrigées de l'absorption avec 12 réflexions mesurées à différents angles ψ (facteurs de transmission de 0,761 à 0,887). R1 = 0,050 pour 4021 réflexions avec I > 2σ(I) et wR2 = 0,135 pour toutes les réflexions, 343 paramètres étant affinés. Les atomes d'hydrogène ont été placés en positions calculées. Le pic résiduel le plus élevé dans la dernière carte de Fourier différence est de 0,385 e/Å3.
2.3 Spectroscopie infrarouge
Les spectres infrarouges (IR) ont été enregistrés sur un spectrophotomètre FT-IR Nicolet Magna 750, entre 3600 et 400/cm avec une résolution de 4/cm. Les échantillons sont dilués dans KBr (ca. 1 %) et pressés pour former des pastilles de 3 mm de diamètre. Les intensités sont abrégées comme suit : TF = très forte, F = forte, m = moyenne, f = faible.
TTF (ν, cm–1) : 3063 (m), 1526 (m), 1253 (F), 1076 (f), 795 (TF), 779 (TF), 734 (m), 649 (TF), 632 (TF) 434 (m). L'assignation des modes de vibration pour ce composé a été publiée [19].
TNF (ν, cm–1) : 3095 (F, νCH), 1734 (TF, νCO), 1617 (F, νC = C), 1598 (F, νC = C), 1542 (TF, νC = C), 1525 (TF, νasNO2), 1447 (m, νC = C), 1344 (TF, νsNO2).
α-(TTF)3(TNF)2 (ν, cm–1) : 3101 (m, νCH), 1734 (F, νCO), 1618 (m, νC = C), 1597 (m, νC = C), 1539 (F, νC = C), 1523 (F, νasNO2), 1342 (TF, νsNO2).
β-(TTF)3(TNF)2 (ν, cm–1) : 3090 (m, νCH), 1727 (F, νCO), 1615 (m, νC = C), 1594 (m, νC = C), 1541 (F, νC = C), 1517 (F, νasNO2), 1340 (TF, νsNO2).
3 Discussion des résultats
3.1 Structures cristallines
Le polymorphe α est une phase triclinique cristallisant dans le groupe d'espace P. L'unité asymétrique est constituée d'une molécule de TTF [S20...S26, désignée comme TTF(1)] et d'une molécule de TNF en positions générales, ainsi que d'une demi-molécule de TTF [S30...C34, désignée comme TTF(2)] proche d'un centre d'inversion. L'unité asymétrique peut donc être formulée AD3/2, ce qui correspond, pour la maille, à la formule minimum A2D3 (Fig. 2). Cette stœchiométrie est peu commune pour les complexes à transfert de charge construits sur le TTF, où les composés de formule AD dominent largement. Quelques complexes organiques de ce type ont cependant été caractérisés par diffraction des rayons X [20–22].
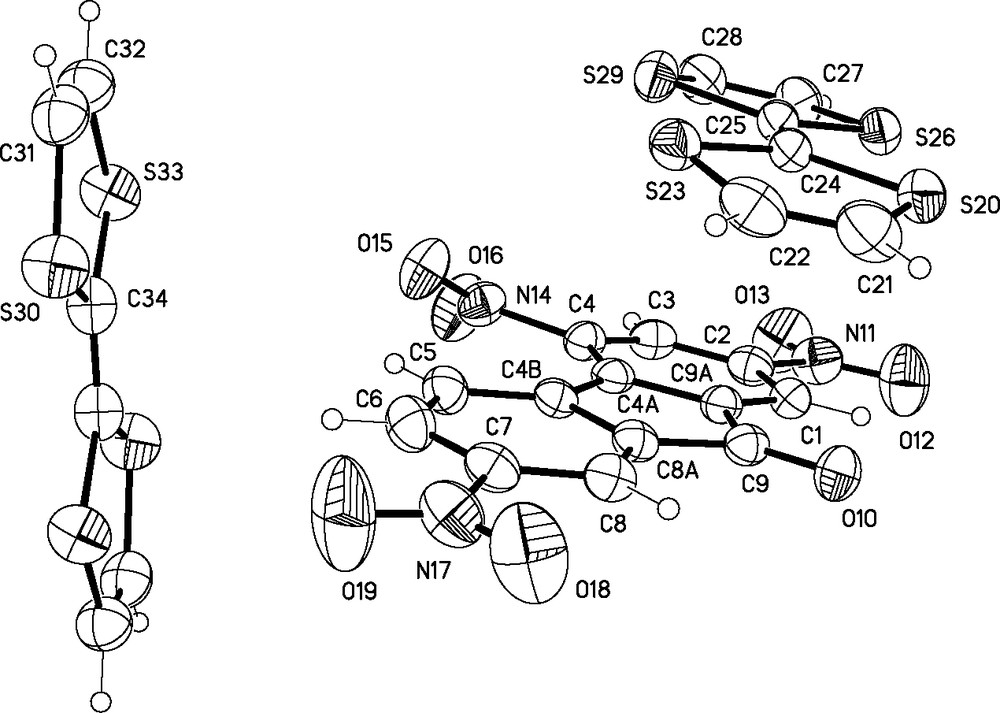
Structure cristalline de α-(TTF)3(TNF)2 avec les ellipsoïdes thermiques à 30 % de probabilité. Le schéma de numération est indiqué pour l'unité asymétrique.
L'étude de l'empilement des molécules A et D permet de formuler plus précisément ce composé comme (Aδ–)2(Dδ+)2(D). En effet, on peut observer l'empilement de la molécule TTF(1) avec l'accepteur TNF, formant une pile orientée le long de l'axe cristallographique a, alors que l'autre molécule, TTF(2), se trouve en dehors de tout empilement (Fig. 3). Le sous-système (AD)n formant la pile est caractérisé par un angle dièdre entre A y D proche de 0 : l'angle calculé entre les plans moyens pour TTF(1) et TNF, en omettant les atomes d'hydrogène, est de 1,22(3)°. Par ailleurs, la distance entre les centres géométriques de A et D est de 3,8357(15) Å. L'autre donneur, TTF(2), fait un angle dièdre de 77,38(4) avec la TNF. La molécule TTF(2) n'étant pas en interaction avec les autres, elle ne pourra pas participer à un éventuel transport électronique.

Empilement cristallin du complexe α-(TTF)3(TNF)2 vu perpendiculairement à l'axe b de la maille. La distance indiquée correspond à la distance entre deux points fictifs représentant les centres géométriques des molécules TTF(1) et TNF participant à l'empilement.
De la même manière, le polymorphe β, qui cristallise également dans le groupe d'espace P, peut formellement s'écrire (Aδ–)2(Dδ+)2(D). L'unité asymétrique est cette fois constituée d'une molécule de TNF en position générale et de trois demi-molécules de TTF proches de trois centres d'inversion (Fig. 4). Des interactions ont lieu principalement le long de l'empilement (DAD′)n, approximativement parallèle à l'axe réciproque c*, où D et D′ sont deux molécules de TTF cristallographiquement indépendantes, à savoir TTF(2) [S25...C29] et TTF(3) [S30...C34]. Les angles dièdres entre les plans moyens des molécules constituant cette pile sont de 2,17(7) et 2,32(7)°, pour les paires D/A et A/D′, respectivement, et les distances entre plans, calculées en prenant les centres géométriques des molécules, valent 3,7494(4) et 4,1752(5) Å, pour D···A et A···D′, respectivement. Finalement, la molécule TTF(1) [S20...C24] sépare les piles dans le cristal, faisant un angle dièdre de 76,03(4)° avec la molécule de TNF (Fig. 5).
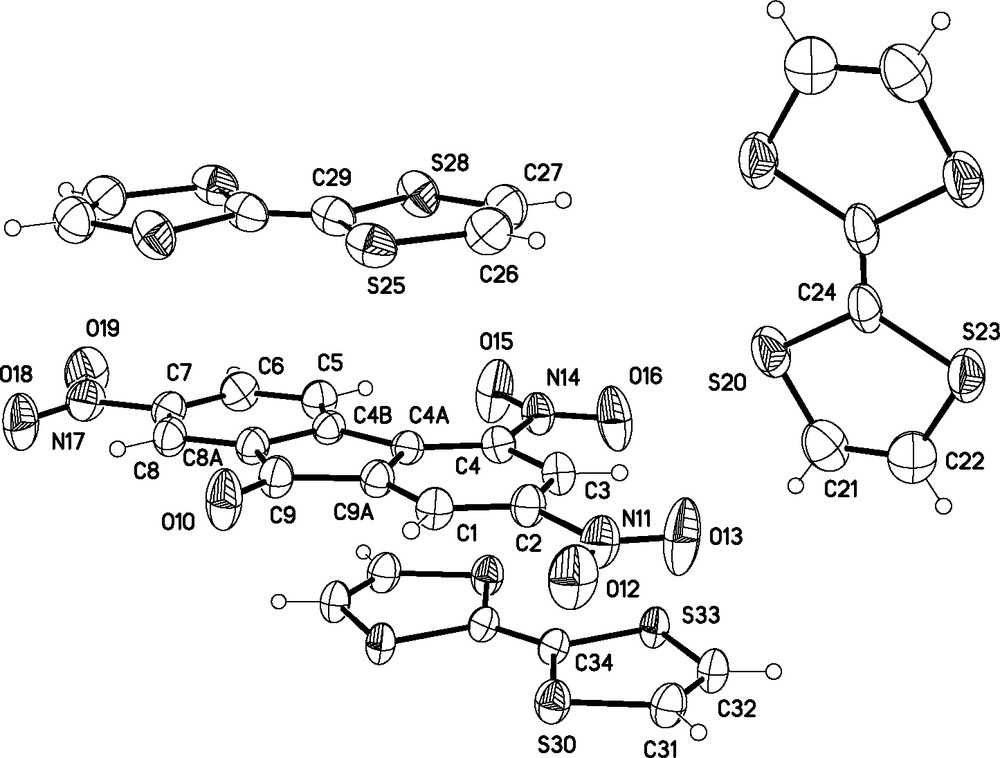
Structure cristalline de β-(TTF)3(TNF)2 avec les ellipsoïdes thermiques à 30 % de probabilité. Le schéma de numération est indiqué pour l'unité asymétrique.

Empilement cristallin du complexe β-(TTF)3(TNF)2 vu perpendiculairement à l'axe a de la maille. Les distances indiquées en traits continus correspondent aux distances entre deux points fictifs représentant les centres géométriques des molécules TTF(2, 3) et TNF participant à l'empilement. La distance indiquée en trait discontinu est un contact intermoléculaire entre un atome d'oxygène de la TNF et un atome de soufre de TTF(3).
Une première question qui se pose est de savoir quel est le phénomène provocant l'apparition de deux polymorphes au sein d'un système homogène, les phases α et β étant obtenues par évaporation lente d'une solution de TTF et TNF (cf. § 2.1). Il semble que la raison principale soit une modification de la géométrie de l'accepteur TNF. Dans le cas du polymorphe α, la molécule de TNF présente le groupe nitro substituant la position 4 de la fluorénone hors du plan de celle-ci. Les plans définissant ce groupe fonctionnel [C4, N14, O15, O16] et le noyau benzène auquel il est attaché [C1, C2, C3, C4, C4a, C9a] forment entre eux un angle de 24,13(9)°. Cette géométrie est proche de celle de la TNF non complexée, pour laquelle le même angle est de 32,7° [23]. Cependant, une situation bien différente est observée pour le polymorphe β, avec un angle de 1,5(2)° pour ce même groupe NO2. La molécule devient alors pratiquement plane : la déviation moyenne hors du plan calculé pour les 23 atomes (C, N et O) est de 0,065 Å. Cette rotation est également visible dans les spectres IR des complexes, avec un déplacement notable des vibrations d'élongation symétrique et asymétrique des groupes NO2 (cf. § 2.3). Cette rotation de l'ordre de 20° d'un groupe nitro représente une demande d'énergie assez faible, inférieure à 1 Kcal/mol selon Dashevskii et al. [24], ce qui rend le phénomène tout à fait accessible aux alentours de 300 K. Cependant, une telle géométrie plane pour la TNF n'avait été observée jusqu'à présent que dans peu de cas [25–27].
Il est clair que cette modification structurale de la TNF oriente l'empilement dans l'état solide. Le volume stérique étant plus faible pour la TNF de géométrie plane, l'empilement pour le polymorphe β présente la distance D···A la plus courte, ca. 3,75 Å et un volume plus faible pour la maille cristalline (cf. § 2.2). Ces différences se reflètent, par exemple, dans les indices d'empilement [28] respectifs : 0,706 pour le polymorphe α, contre 0,710 pour le polymorphe β. Une autre conséquence importante concernant les propriétés de transport est que la TNF plane doit être un meilleur π-accepteur, ayant un système aromatique plus étendu, ce qui doit favoriser le transfert de charge dans la phase β.
3.2 Estimation du degré de transfert de charge
Il a été démontré, de manière empirique, qu'une des conditions nécessaires pour obtenir un complexe donneur-accepteur à caractère métallique est que le degré de transfert de charge δ doit être compris entre 0,4 et 0,7 [29], ce qui est le cas, par exemple, pour le solide moléculaire TTF-TCNQ, où δ = 0,59 [30] (TCNQ est le 7,7,8,8-tétracyanoquinodiméthane). La méthode la plus simple pour estimer δ consiste à étudier les déplacements de fréquence en spectroscopie vibrationnelle (IR et Raman) pour les liaisons intramoléculaires sensibles à la modification de la charge de la molécule due au transfert de charge [29,31]. Un cas classique est l'utilisation de la vibration d'élongation des groupes nitriles dans les complexes du TCNQ [31]. Comme des complexes à transfert nul et total sont disponibles pour cet accepteur, il est possible de calculer δ à partir de la fréquence de vibration νC≡N. La méthodologie peut être transférée à d'autres accepteurs contenant le même groupe fonctionnel et permet d'évaluer δ avec une précision de l'ordre de 0,05 [16].
Dans le cas qui nous occupe, le transfert de charge est probablement de faible intensité. En effet, pour un complexe TTF–TNF, dont la structure cristalline n'est pas connue, δ a été évalué sur la base de spectres Raman : δ ≈ 0,05 [32]. Par ailleurs, les séparations D···A observées dans l'intervalle 3,75–4,17 Å pour (TTF)3(TNF)2 sont relativement grandes comparées, par exemple, à la distance D···A = 3,74 Å pour le complexe 1:1 construit avec TTF et la 2,7-dinitro-9-fluorénone [12], pour lequel δ ≈ 0,09 [32].
La double liaison centrale du TTF est certainement sensible au transfert de charge, pour être une liaison π et pour se situer au centre géométrique de la molécule. D'ailleurs, cet effet est visible dans les structures cristallines : la distance C=C centrale du TTF s'allonge quand cette molécule est oxydée, comparée à la distance observée pour les molécules neutres. Malheureusement, la vibration active en IR pour cette liaison (élongation, mode b1u [19]) est peu intense et, surtout, se superpose à une vibration intense de la TNF, ce qui la rend inutilisable. En revanche, pour l'accepteur, la vibration d'élongation du carbonyle se situe dans une zone où le TTF n'absorbe pas. On constate que la position de cette bande IR reste inchangée pour α-(TTF)3(TNF)2 par rapport à TNF, ce qui coïncide avec un transfert de charge δ ≈ 0, alors que pour β-(TTF)3(TNF)2 un déplacement significatif est observé, Δν = 7/cm, qui doit correspondre à δβ > 0. Ce résultat qualitatif (δβ > δα) est en accord avec les observations antérieures : le polymorphe β contient une molécule de TNF plane, meilleur π-accepteur que la TNF non complexée, ainsi qu'une distance D···A plus courte (3,75 Å) que pour le polymorphe α. Une autre caractéristique structurale peut expliquer pourquoi δβ > δα : un contact intermoléculaire entre le TTF et la TNF de deux piles voisines est observé dans le cristal de β-(TTF)3(TNF)2, mettant en jeu un atome de soufre et l'atome d'oxygène du carbonyle de l'accepteur : O10···S33i = 2,985(2) Å (i est l'opérateur de symétrie 1–x, 1–y, 1–z). Une telle distance, 0,34 Å plus courte que la somme des rayons de van der Waals, n'est pas observée pour la phase α, dans laquelle une molécule de TTF se trouve toujours en face d'une autre molécule de TTF de la pile voisine, et de même pour la TNF (Fig. 3).
Quant aux propriétés de conduction, on peut supposer a priori que ces complexes sont isolants. En effet, l'une des méthodes empiriques qui permettent d'évaluer les chances qu'a un adduit de présenter une conductivité électrique notable est de comparer les potentiels de première oxydation du donneur (E1D) et de première réduction de l'accepteur (E1A). Wheland a montré [33,34] que pour un couple constitué d'un donneur modéré (0,1 < E1D < 0,4 V) et d'un accepteur modéré (–0,02 < E1A < 0,34 V), la probabilité de former un complexe conducteur est élevée si |E1A – E1D| < 0,25 V. Or, le potentiel de réduction de la TNF dans l'acétonitrile est E1A = –0,46 V [35], tandis le TTF s'oxyde dans le même solvant à E1D = +0,34 V [36]. L'écart |E1A – E1D| = 0,8 V est donc très au-delà du seuil empiriquement déterminé par Wheland. Par ailleurs, le fait que les deux phases montrent des empilements alternés DA n'est pas favorable, la majorité des conducteurs moléculaires étant fondés sur des architectures d'empilements ségrégués. Il convient cependant de signaler que des résultats inattendus sont parfois obtenus avec ce type de complexes, un exemple récent étant la synthèse du premier conducteur métallique unimoléculaire neutre [37,38].
4 Conclusions
Nous avons isolé deux phases polymorphes pour le complexe (TTF)3(TNF)2 et montré que la spectroscopie IR permet d'évaluer le degré de transfert de charge, alors même que celui-ci est faible. Une question qui reste en suspens concerne le paramètre expérimental qui permet d'orienter la cristallisation vers une forme ou une autre. Apparemment, la concentration durant le processus de nucléation paraît cruciale, mais nous ne savons pas s'il s'établit un équilibre entre les phases α et β en solution, ou bien si une transformation de phase α → β a lieu durant la cristallisation. Par ailleurs il n'est pas impossible que d'autres stœchiométries soient accessibles pour le système TTF–TNF en solution, qui pourraient permettre la préparation de nouvelles phases cristallines, stables ou métastables.
5 Matériel supplémentaire
Le matériel supplémentaire pour les structures cristallines a été déposé au Cambridge Crystallographic Data Center sous forme de fichiers CIF, et peut être obtenu auprès du CCDC (www.ccdc.cam.ac.uk) en citant les numéros de dépôt : CCDC-244794 (phase α) et CCDC-244795 (phase β). Les intensités de diffraction sont disponibles auprès des auteurs.