

1 Introduction
Sugars, especially D-glucose and D-fructose, are essential energy sources for mammalian cells. It is known that these hexoses are incorporated into cells by a facilitated diffusion process through the plasma membrane mediated by a family of transmembranar proteins named GLUT [1]. Thirteen isoforms of these proteins (GLUT1-13) are already known [2], GLUT5 being a fructose-specific transporter [3]. Previous structural inhibition studies have shown that fructose is recognized by its transporter under both pyranose and furanose forms [4]. Furthermore, it was shown that the 6-OH group of the furanose form is a non-essential position in this recognition phenomenon and could therefore be chemically modified for the elaboration of GLUT5 inhibitors [5]. Such inhibitors would be valuable labelling tools for better understanding of D-fructose uptake into cells and for the quantification of the membranar density of GLUT5 protein. As a usual feature, inhibition constants for the uptake by GLUT5 are found to be in the millimolar range (Ki # 15mM for D-fructose; [4]), which is quite usual for protein-sugar recognition processes.
In order to obtain more efficient probes and following some previous works in the D-glucose series [6,7], our initial strategy was based on the elaboration of multivalent inhibitors for GLUT5. Indeed, it is well known that great binding enhancements in protein-carbohydrate recognition processes could be observed when comparing monomeric to multivalent sugar-based ligands [8]. Though, our choice was brought on the synthesis of multivalent glycopeptides containing the proposed fructose-based recognition pattern. Three fructose-like compounds were choosen in order to mimic the recognition of D-fructose by GLUT5: 1-amino-1-deoxy-2,5-anhydro-D-mannitol 1, 2,5-dideoxy-2,5-imino-D-mannitol 2 and 6-amino-6-deoxy-α,β-D-fructofuranose 3 (Fig. 1). Compounds 1 and 2 are amino analogues of 2,5-anhydro-D-mannitol 4, which presents a slightly better inhibitory effect than D-fructose itself (Ki # 13mM; see [4]), while 3 is the amino analogue of α,β-D-fructofuranose 5 (Fig. 1). Furthermore, some preliminary biological evaluations of the C-2 symmetrical polyhydroxylated pyrrolidine 2 have shown that its affinity for GLUT5 is still in the millimolar range (IC50 # 20mM). Thereby, this molecule is a good synthetic starting point for the elaboration of high-affinity GLUT5 inhibitors.
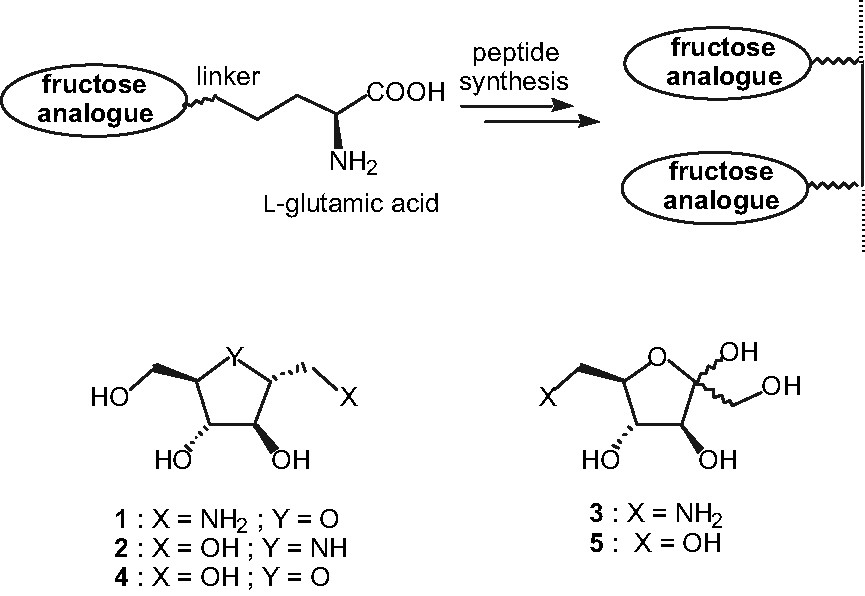
General synthetic strategy for the amplification of fructose recognition pattern.
The amplification step will be done by peptide synthesis with a terminal α-amino acid moiety attached to the sugar via a well-adapted linker. As it would allow for selective functionalizations from its side-chain, L-glutamic acid 6 appears to be a suitable substrate for this purpose (Fig. 1).
In this paper, we will describe the preparation of five glycopeptides by conjugating these D-fructofuranose analogues 1-3 and L-glutamic acid derivatives in order to evaluate their affinity as monomeric ligands for GLUT5 before further amplification.
2 Results and discussion
The first synthetic target was glycoconjugate 14 bearing a free α-amino acid moiety on a single pseudo-sugar unit (Fig. 2). We first plan to link these moieties by an amino linker obtain via reductive amination using the protected amine 10. This primary amine had already been obtained by hydrogenolysis of acetoxime 8 [9] (Fig. 2).
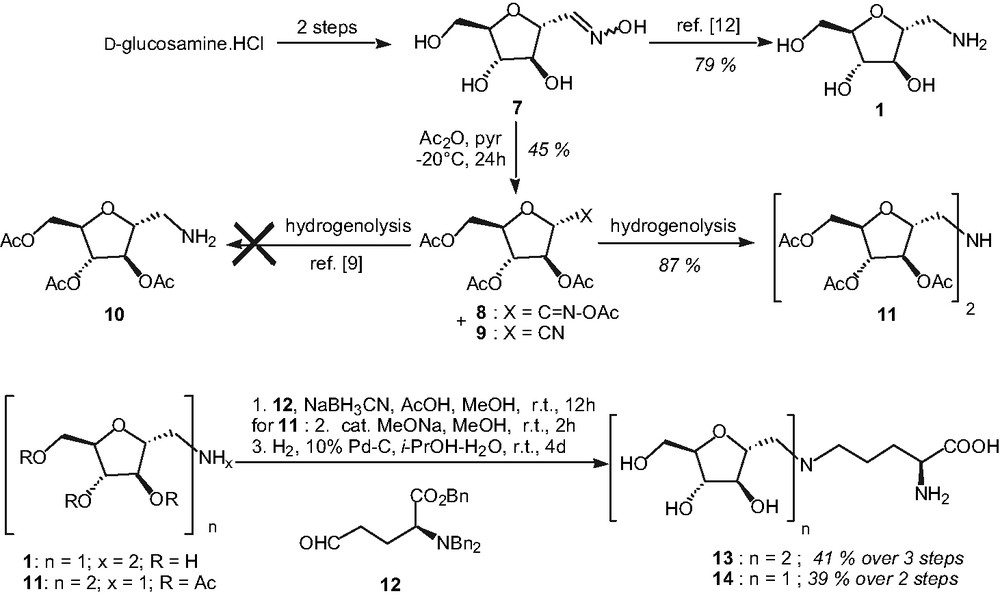
Syntheses of glycopeptides 13 and 14 with an amine linker.
However, in our hands, hydrogenolysis of either acetoxime 8 or nitrile 9 (both compounds were formed during the acetylation step; [9]) led to the exclusive formation of secondary amine 11 in good yield (Fig. 2). This feature was proved by DCI mass spectrometry (ion [11+H]+ at m/z 562 consistent with gross formula C24H35NO14 for 11) and by examination of the relative integrations of 1H NMR signals of the N-acetylated derivatives in both cases (Ac2O, pyridine). The formation of such secondary amines from oximes or nitriles can be explained by a dimerization process with loss of ammonia and has already been observed under hydrogenolytic conditions [10]. Still, it was decided to take advantage of this unexpected result, and amine 11 was engaged in a reductive amination reaction with aldehyde 12 (obtained from 6 in two steps; [11]). After a two steps deprotection sequence, compound 13 bearing two pseudo-sugar units and one α-amino acid unit has been obtained in a 41% yield over three steps (Fig. 2). Nevertheless, we were still interested in the preparation of a glycoconjugate containing a single pseudo-sugar unit. This one was obtained from the fully unprotected compound 1 prepared from oxime 7 [12]. After reductive amination in the presence of aldehyde 12 followed by complete hydrogenolysis of the benzyl groups, 14 was isolated in 39% yield (Fig. 2).
We also decided to build three other model compounds (15, 16 and 17) containing a pseudo-fructose unit linked to a L-glutamic acid derivative via an amide bond (Fig. 3). The syntheses of 15 and 16 were accomplished by performing peptide coupling reactions between unprotected pseudo-sugars 1 and 2 (synthesized in six steps from L-sorbose; see [13]) and a suitable L-glutamic acid peptide 18 (Fig. 3). Compound 18 was previously prepared in a three steps procedure from L-glutamic acid γ-methyl ester (1. Ac2O, aq. NaHCO3, dioxane-H2O, r.t., 12h; 2. DCC, pentafluorophenol, AcOEt, r.t., 2h then n-propylamine, DIEA, CH2Cl2, r.t., 2h; 3. 1 N aq. NaOH, THF, 0°C, 0.5h then H3O+). For the coupling step, use of DCC/HOBt/DIEA failed in both cases, leading to complex reaction mixtures in which only pyroglutamic compounds could be identified by NMR. These problems were solved using PyBop® as a coupling reagent [14], leading to the formation of the desired adducts 15 and 16 in moderate but satisfying isolated yields considering our unprotected poly-functionnalized substrates (Fig. 3).

Syntheses of glycopeptides 15,16 with an amide linker.
Both compounds were purified by preparative reversed-phase HPLC: on one side, this purification afforded pure 15. In the case of 16, an unseparable mixture of peptide 16 together with esters such as 19 was obtained (Fig. 3). These esters (19) were cleaved under transesterification reaction conditions and final purification by preparative reversed-phase HPLC produced pure 16. Finally, the third glycopeptide 17 was prepared in four steps sequence from methyl 6-azido-6-deoxy-1,3,4-tetra-O-acetyl-α,β-D-fructofuranoside 20 [15] via coupling with pentafluorophenyl ester 21 and classical deprotection steps (Fig. 3). In this case, the use of an anomerically blocked sugar was essential in order to avoid the possible formation of a polyhydroxylated piperidine during the hydrogenolysis of the 6-azido group as previously described [15,16].
In conclusion, we have investigated the syntheses of new potential inhibitors of GLUT5 protein, a fructose-specific transporter. In order to obtain high-affinity GLUT5 multivalent inhibitors by amplification of the D-fructofuranose recognition pattern, five model compounds have been prepared from D-fructofuranose amino analogues 1–3 and derivatives of L-glutamic acid 6. The former (13, 14) bear a free α-amino acid moiety through an amine linker and were prepared by a sequence involving aldehyde 12 in a reductive amination step. The later (15–17) possessing an amide linker have been prepared via peptide coupling with L-glutamic acid derivatives 18 and 21. Compounds 15–17 are now under biological evaluations from which results will be reported in due course. Depending on their relative affinities for GLUT5 compared to that of D-fructose, they will be considered for further amplification steps by peptide synthesis. This will be done using analogues suitably functionalized on the amino acid moiety for peptide synthesis strategy.
3 Experimental Section
Methanol was distilled over magnesium, tetrahydrofuran was distilled over sodium-benzophenone, dichloromethane was distilled over calcium hydride, DMF was distilled over 4-Å molecular sieves. Analytical TLC was carried out on precoated Merck silica gel 60F-254 plates, visualized under UV light (λ = 254 nm) and developed by the use of either an ethanolic solution of ninhydrin or an alkaline aqueous solution of potassium permanganate. Purifications by column chromatography were performed on E. Merck flash silica gel SI 60 (43–60 μm). Purifications by preparative reversed-phase high performance liquid chromatography (RP-HPLC) were performed on a Waters equipment using a C18 column (Nucleosil 300 μ; 250 × 21 mm) monitored at λ = 214 nm using the following A-B gradient: 5 to 40% B in 30 min with a 18-ml/min debit (A: 0.09% trifluoroacetic acid in water; B: 0.09% trifluoracetic acid in a (9:1) mixture of acetonitrile–water). All evaporations of volatiles were performed at t < 30 °C under reduced pressure except for water which was removed by lyophilisation. 1H and 13C spectra were recorded on a Bruker Advance 300 MHz spectrometer. ESI-mass spectra were measured in the positive mode on a VG Platform II. DCI-mass spectra (NH3/isobutane) were measured on a Thermofinnigan Polaris Q in the positive mode. HRMS analyses were performed at the CRMPO, University of Rennes-1.
3.1 Syntheses of compounds 13 and 14
3.1.1 Compound 13
To a solution containing amine 11 (75 mg; 0.134 mmol) and aldehyde 12 (54 mg; 0.134 mmol) in dry methanol (2.5 ml) under argon were sequentially added sodium cyanoborohydride (25 mg; 0.401 mmol) and glacial acetic acid (8 μl; 0.140 mmol). The mixture was stirred overnight and solvents were evaporated under reduced pressure. The resulting slurry was diluted with ethyl acetate (5 ml), the organic layer was washed with a saturated aqueous solution of sodium hydrogenocarbonate (2 × 5 ml), and the aqueous layer was extracted with ethyl acetate (2 × 5 ml). The combined organic layers were washed with brine (10 ml) and dried over anhydrous sodium sulphate. After evaporation of volatiles under reduced pressure, the crude product was purified by flash chromatography (eluent: n-pentane-ethyl acetate (2:1); Rf # 0.35) to give 63 mg (50%) of a colorless oil which was then dissolved under argon in dry methanol (2.0 ml). At room temperature, one drop of a 6 N solution of sodium methoxide in methanol was added and the mixture was stirred for 2 h. One drop of a 6 N aqueous solution of hydrochloric acid was then added and the solvent was removed under reduced pressure. The resulting slurry was dissolved in 2.0 ml of absolute ethanol and filtrated through a short pad of celite. The filtrate was concentrated under reduced pressure and purified by flash chromatography (sequential eluents: 1, ethyl acetate; 2, ethyl acetate-methanol (9:1); 3, absolute ethanol; Rf # 0.5 in absolute ethanol) affording 43 mg (93%) of a colorless oil. This product was then dissolved in a (2:1) mixture of iso-propanol and water (1.5 ml). 10 mg of 10% palladium on carbon was added and hydrogenolysis was carried out at atmospheric pressure and room temperature for 4 days. After filtration on a short pad of celite and removal of solvents under reduced pressure, compound 13 (23 mg; 0.054 mmol; 41% overall isolated yield over 3 steps) was obtained as a pale yellow oil. 1H NMR (D2O, 300 MHz): 4.17–3.96 (m, 8H, H-2, H-3, H-4 and H-5 of sugar units), 3.87–3.71 (m, 5H, H-6 of sugar units and CHN), 3.12–2.84 (m, 6H, H-1 of sugar units and NCH2CH2CH2), 1.98–1.93 (m, 2H, NCH2CH2CH2), 1.76–1.72 (m, 2H, NCH2CH2CH2); 13C NMR (D2O, 75 MHz): 83.0, 79.6, 79.3 and 76.9 (C-2, C-3, C-4 and C-5 of sugar units), 61.7 (C-6 of sugar units), 56.6 (C-1 of sugar units), 54.6 (CHN), 54.3 (NCH2CH2CH2), 28.9 (NCH2CH2CH2), 21.3 (NCH2CH2CH2); ESI-MS (m/z): 447 [M+Na]+.
3.1.2 Compound 14
Same procedure for the reductive amination step using 175 mg (0.605 mmol) of 1, 243 mg (0.605 mmol) of aldehyde 12, 115 mg (1.817 mmol) of sodium cyanoborohydride, 35 μl (0.605 mmol) of glacial acetic acid in 55 ml of dry methanol. Purification by flash chromatography on silica gel treated with eluents containing 0.5% triethylamine (sequential eluents: 1, ethyl acetate; 2, ethyl acetate-methanol (95:5); 3, absolute ethanol ; Rf # 0.3 in absolute ethanol) afforded 160 mg (40%) of the desired product. Hydrogenolysis was accomplished at room temperature for 4 days under an atmospheric pressure of hydrogen using 25 mg of 10% palladium on carbon in a (9:1) mixture of iso-propanol and water (3.0 ml). This gave access to compound 14 (61 mg; 0.233 mmol; 39% over 2 steps) as a pale yellow oil. 1H NMR (D2O, 300 MHz): 4.19-3.92 (m, 4H, H-2, H-3, H-4 and H-5 of sugar unit), 3.85–3.69 (m, 3H, H-6 of sugar unit and CHN), 3.21 (d, 2H, J = 8.3Hz, H-1 of sugar unit); 3.09–3.02 (m, 2H, NCH2CH2CH2), 1.94–1.65 (m, 4H, NCH2CH2CH2); 13C NMR (D2O, 75 MHz): 83.4, 79.4, 79.0 and 77.0 (C-2, C-3, C-4 and C-5 of sugar unit), 61.6 (C-6 of sugar unit), 54.8 (CHN), 49.7 and 47.9 (C-1 of sugar unit, NCH2CH2CH2), 28.9 (NCH2CH2CH2), 23.0 (NCH2CH2CH2); ESI-MS (m/z): 285 [M+Na]+.
3.2 Syntheses of compounds 15-17
3.2.1 Compound 15
To a solution of amine 1 (25 mg; 0.153 mmol) and acid 18 (35 mg; 0.153 mmol) in dry DMF (1.0 ml) were sequentially added under argon atmosphere N,N-diisopropylethylamine (135 μl; 0.775 mmol) and PyBop® (80 mg; 0.153 mmol). The mixture was stirred at room temperature until complete disappearance of any ninhydrin active compound on TLC (eluent: absolute ethanol-22% aqueous ammonia (9:1)). The reaction was completed in 30 min, volatiles were removed under reduced pressure and the resulting slurry was triturated with dichloromethane (3 × 5 ml). The resulting solid was then dissolved in water and purified by preparative reversed-phase HPLC (tR = 7.5 min) to afford 15.5 mg (28%) of 15 as a pale yellow oil. 1H NMR (D2O, 300 MHz): 4.26–3.51 (m, 4H, H of sugar unit and CHN), 3.20 (t, 2H, J = 6.2 Hz, NCH2CH2CH3); 2.42–2.38 (m, 2H, NHCOCH2CH2), 2.08-1.99 (m containing s at 2.04ppm, 5H, NHCOCH2CH2 and CH3 acetamide), 1.58–1.51 (m, 2H, NCH2CH2CH3); 0.91 (t, 3H, J = 7.0 Hz, NCH2CH2CH3); 13C NMR (D2O, 75 MHz): 175.8, 174.8 and 173.9 (C=O amides), 83.2, 81.2, 78.6 and 77.1 (C-2, C-3, C-4 and C-5 of sugar unit), 61.8 (C-6 of sugar unit), 54.0 (CHN), 41.9 and 41.4 (C-1 of sugar unit, NHCH2CH2CH3), 32.5 (NHCOCH2CH2), 27.8 (NHCOCH2CH2), 22.4 (NCH2CH2CH3), 22.3 (CH3 acetamide), 11.1 (NCH2CH2CH3); ESI-MS (m/z): 398 [M+Na]+, 376 [M+H]+; ESI-HRMS calcd for C16H29N3O7Na: 398.1903 [M+Na]+, found: 398.1904.
3.2.2 Compound 16
Same procedure for the peptide coupling than described above using 25 mg (0.153 mmol) of 2 and the same amounts of the other reagents. Reaction was monitored by TLC (eluent: MeOH-22% aqueous ammonia (7:3)) following the disappearance of amine 2 (Rf # 0.65) revealed by an ethanolic solution of ninhydrin. In this case, reaction was completed in 30 min. Purifcation by preparative reversed-phase HPLC (tR = 8.9 min) gave an unseparable mixture of 16 together with esters 19. This mixture was then treated in dry methanol (500 μl) by a catalytic amount of a 1 N solution of sodium methoxide in methanol. After 1 h at room temperature, volatiles were removed under reduced pressure, the residue was then dissolved in water (1.5 ml) and treated with ion-exchange resin Dowex 50WX8 (H+ form) until pH 7. After filtration, lyophilisation and purification by reversed-phase HPLC (tR = 8.9 min) afforded 11.5 mg (0.030 mmol; 20% over 2 steps) of pure 16 as a colorless oil. 1H NMR (D2O, 300 MHz): 4.30–4.28 (m, 2H, H-3 and H-4 of sugar unit), 4.08–4.02 (m, 2H, H-2 and H-5 of sugar unit), 3.98–3.82 (m, 5H, H-1 and H-6 of sugar unit and CHN), 3.25–3.20 (m, 2H, NCH2CH2CH3); 2.66-2.61 (m, 2H, NHCOCH2CH2), 2.24–1.97 (m containing s at 2.10 ppm, 5H, NHCOCH2CH2 and CH3 acetamide), 1.62–1.50 (m, 2H, NCH2CH2CH3); 0.93 (t, 3H, J = 7.4 Hz, NCH2CH2CH3); 13C NMR (D2O, 75 MHz): 175.2, 174.6 and 173.8 (C=O amides), 77.9 and 77.8 (C-3 and C-4, 2 rotamers), 68.6 and 68.5 (C-2 and C-5 of sugar unit, 2 rotamers), 61.6 and 59.6 (C-1 and C-6, 2 rotamers), 61.8 (C-6 of sugar unit), 54.1 (CHN), 41.5 (NHCH2CH2CH3), 31.1 (NHCOCH2CH2), 27.4 (NHCOCH2CH2), 22.2 (CH3 acetamide), 22.1 (NCH2CH2CH3), 10.9 (NCH2CH2CH3); ESI-MS (m/z): 398 [M+Na]+, 376 [M+H]+; ESI-HRMS calcd for C16H29N3O7Na: 398.1903 [M+Na]+, found: 398.1904.
3.2.3 Compound 17
Azide 20 (236 mg; 0.683 mmol) was dissolved in absolute ethanol (5.0 ml) and 25 mg of 10% palladium on carbon were added. The hydrogenolysis was carried out under an atmospheric pressure of hydrogen at room temperature for 20 h. After filtration on a celite pad and evaporation of the solvent under vacuo, 198 mg (0.624 mmol; 91%) of the corresponding amine were obtained as a colorless oil. 51 mg (0.163 mmol) of this compound were dissolved in dry dichloromethane (1.5 ml) under an argon atmosphere. N,N-diisopropylethylamine (85 μl; 0.489 mmol) and pentafluorophenyl ester 21 (75 mg; 0.189 mmol) were then sequentially added under argon atmosphere. The mixture was stirred at room temperature until complete disappearance of the amine occurred (monitored by TLC; eluent: ethyl acetate; Rf amine # 0.05). After 12h, solvent was removed under vacuo and the crude mixture was purified by on flash silica gel column chromatography (eluent: ethyl acetate-methanol (3:1); Rf of coupled product # 0.60) to afford 65 mg (0.122 mmol; 63%) of the desired coupling product. Acetates were removed using a catalytic amount of sodium methoxide in anhydrous methanol (1.0 ml), the reaction being monitored by TLC (eluent: ethyl acetate–methanol (3:1); Rf deacetylated compound # 0.10). After stirring at room temperature for 3 h, reaction was complete and usual work-up procedure was performed affording the desired deacteylated product as a brown viscous oil (50 mg), which was then dissolved in water (2.0 ml) and treated with ion-exchange resin Dowex 50WX8 (H+ form) (100 mg) and stirred at room temperature for 1 day. After filtration and lyophilisation, crude 17 was purified by preparative reversed-phase HPLC (tR = 8.1 min) to afford 14.0 mg (0,036 mmol; 20% over 4 steps) as a colorless oil. 1H NMR (D2O, 300 MHz): 4.31-3.85 (m, 4H, H-3, H-4, H-5 of α- and β-D-sugar units and CHN), 3.64–3.60 (m, 2H, H-1 of α− and β-D-sugar units), 3.56–3.50 (m, 2H, H-6 of α- and β-D-sugar units), 3.27–3.15 (m, 2H, NCH2CH2CH3); 2.46–2.37 (m, 2H, NHCOCH2CH2), 2.23-1.95 (m containing s at 2.09ppm, 5H, NHCOCH2CH2 and CH3 acetamide), 1.61–1.49 (m, 2H, NCH2CH2CH3); 0.93 (m, 3H, NCH2CH2CH3); 13C NMR (D2O, 75 MHz): 175.6, 174.7 and 173.7 (C=O amides), 104.9 and 102.0 (C-2 of α- and β-D-sugar units), 79.7, 79.0, 77.8, 76.2, 76.1 and 75.7 (C-3, C-4 and C-5 of α- and β-D-sugar units), 63.2 and 63.1 (C-1 of α- and β-D-sugar units), 54.0 (CHN), 42.2, 41.5 and 41.1 (C-6 of α- and β-D-sugar units and NHCH2CH2CH3), 32.3 (NHCOCH2CH2), 27.5 (NHCOCH2CH2), 22.2 (NCH2CH2CH3), 22.1 (CH3 acetamide), 10.9 (NCH2CH2CH3); ESI-MS (m/z): 414 [M+Na]+, 392 [M+H]+, 374 [M+H-H2O]+; ESI-HRMS calcd for C16H29N3O8Na: 414.1854 [M+Na]+, found: 414.1852.
Acknowledgments
The author would like to thank Dr. T. Joët and Prof. S. Krishna (St George's Hospital, London, UK) for the biological evaluations on CHO cells over-expressing GLUT5.