1 Introduction
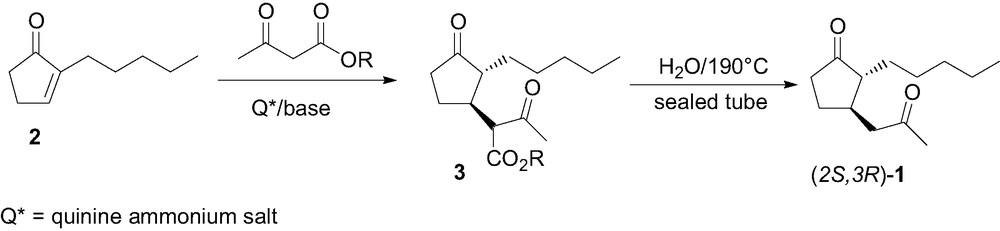
Many floral, jasmine-like fragrances, very important for the perfume industry, possess a 2,3-disubstituted cyclopentanone backbone [1]. In the last few years several stereoselective syntheses of methyl dihydrojasmonate (Fig. 1), the most popular cyclopentanone fragrance, have been reported [2], allowing to evaluate the olfactory properties of its single stereoisomers. It was then observed that in jasmonates the olfactory active stereoisomers have (R) stereochemistry at C(3), with the cis stereoisomer being more active than the trans one [2c,3]. A structurally related cyclopentanone fragrance is magnolione (1), in which the substitution of the methyloxycarbonyl moiety of jasmonates with an acetyl group confers a greater odor strength, a better stability, and a more floral, intense jasminic note [1c]. Despite its industrial importance, until now 1 has been prepared and used in the perfume and cosmetic industry as a racemic mixture of cis/trans stereoisomers [4]. Therefore the correlation between its olfactory properties and stereostructure is not known. The knowledge of such correlation is very important since in many chiral fragrances, as seen in the jasmonates, the olfactory properties of the several stereoisomers differ significantly. We then faced the diastereo- and enantioselective synthesis of both enantiomers of trans-1 with the scope to establish its structure/odor relationship. The use of phase transfer catalysis (PTC), recently applied to the enantioselective syntheses of related trans-dihydrojasmonates [2d], looked very appealing also for our purposes. The PTC approach requires, in fact, simple experimental procedures and very mild conditions, which make this process easy to scale-up and interesting for industrial preparation [5,6]. The synthesis of trans-1 was then approached (Scheme 1) by Michael addition of ethyl acetoacetate to 2-pentyl-2-cyclopentenone (2) under PTC conditions in the presence of chiral ammonium salts, leading, after hydrolysis and decarboxylation of the addition product 3, to trans-1 with moderate to good diastereo- and enantio-selectivity [7]. We decided then to extend our previous investigation using other acetoacetic esters, other enantiopure ammonium salts, and employing different experimental conditions. We report herein the results of this full study on the asymmetric PTC synthesis of cyclopentanone fragrance trans-1 with the aim to improve the diastereo- and enantio-selectivity of the reaction.

Fragrances methyl dihydrojasmonate and magnolione (1).
2 Results and discussion
The conjugate addition of ethyl or tert-butyl acetoacetate to enone 2 under solid/liquid PTC conditions, in the presence of a catalytic amount of enantiopure quaternary ammonium salts, was then carried out. We tested several reaction conditions (solvent, base, acetoacetic ester) and the efficiency of different chiral ammonium salts (Fig. 2). We then decided to test N-9-anthracenylmethylquininium chloride (4) [8] and N-9-anthracenymethylquinidinium chloride (5) [8], i.e. the ammonium salts employed in the PTC synthesis of jasmonates [2d] and, for the first time in this reaction, the two 1,1′-binaphthylazepine salts (S,S)-6 e (S)-7. Our group had in fact a large experience in the synthesis of 1,1′-binaphthylazepine chiral ligands and in their use in asymmetric catalysis [9], therefore the use of 1,1′-binaphthylazepine chiral salts, successfully employed in asymmetric synthesis of aminoacids by PTC alkylation [10], seemed very intriguing.
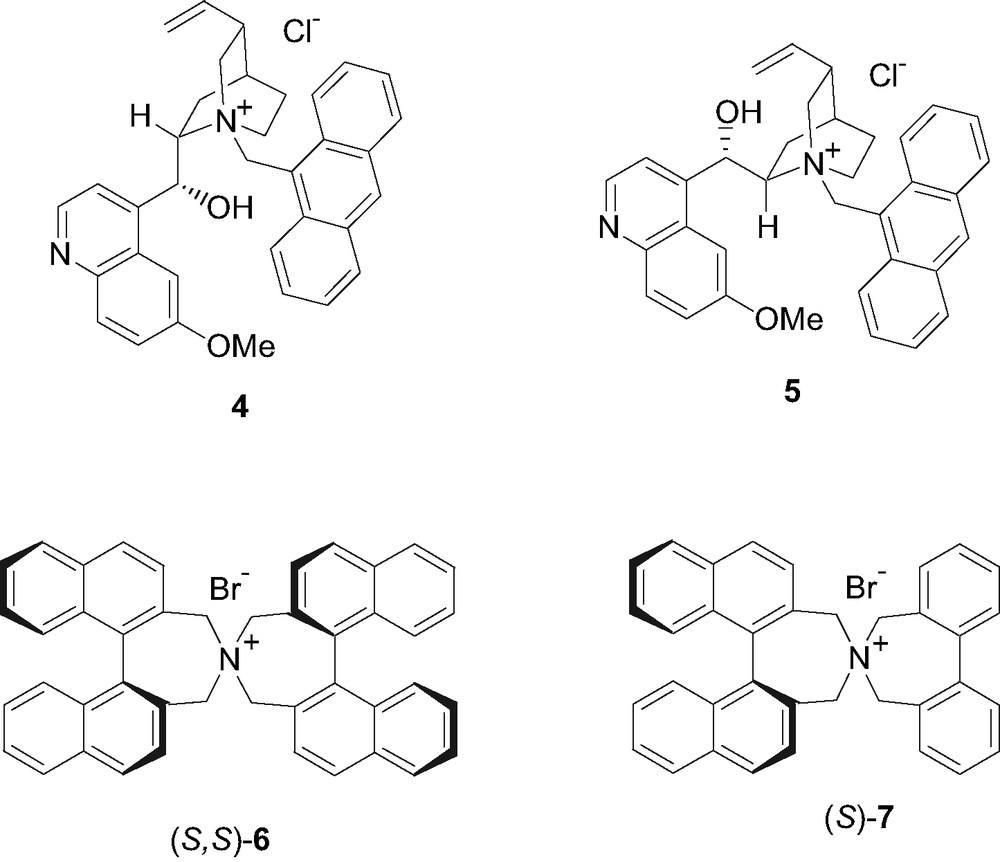
Cinchona-derived ammonium salts 4, and 5. 1,1′-Binaphthylazepine ammonium salts (S,S)-6 and (S)-7.
2.1 Synthesis of chiral ammonium salts
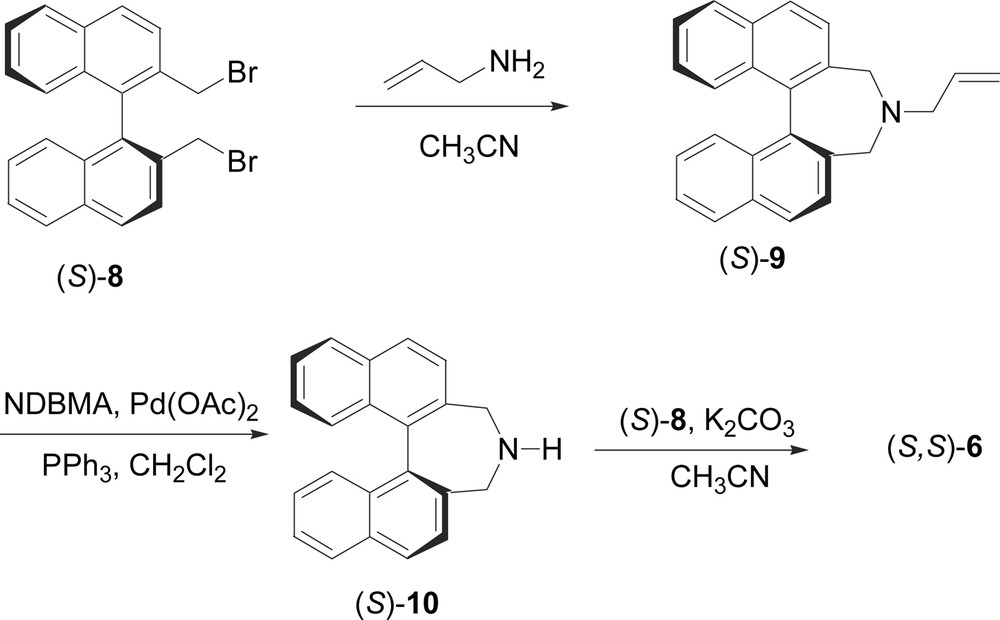
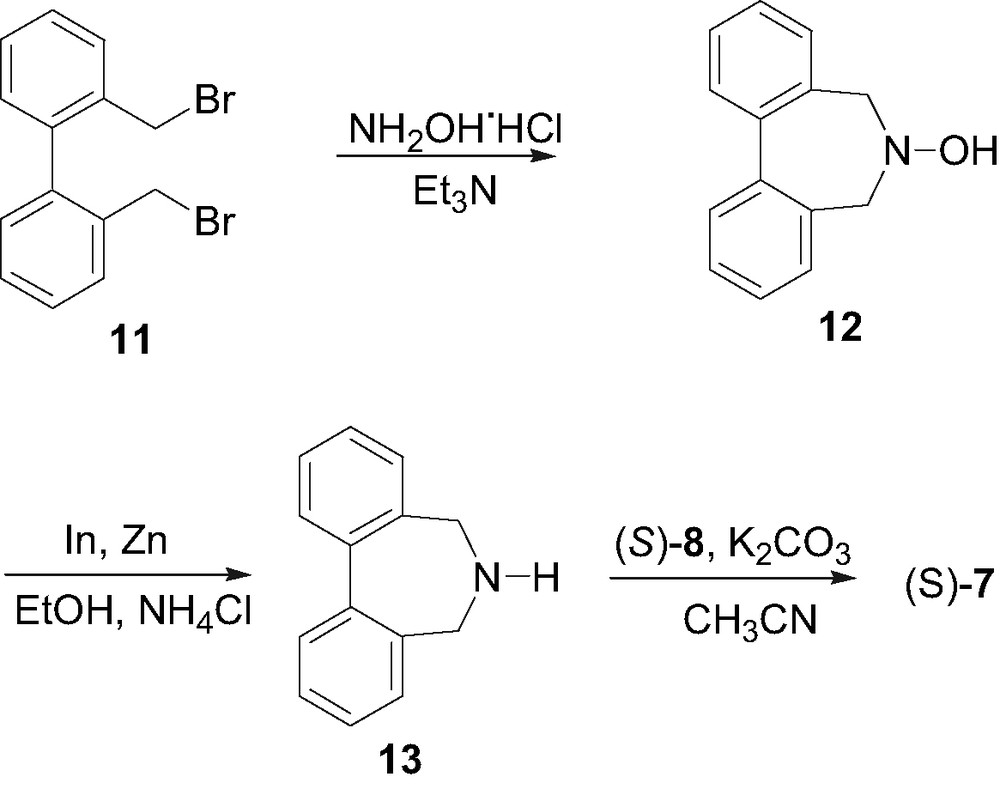
The salts 4 and 5 were obtained by simple alkylation of the parent Cinchona alkaloids, quinine and quinidine, with 9-chloromethylanthracene [2d]. The 1,1′-binaphthylazepine salt (S,S)-6 was obtained following the procedure described by Maruoka et al. [11] starting from (S)-2,2′-bis(bromomethyl)-1,1′-binaphthalene (8) (Scheme 2) [12,9a]. The dibromide (S)-8 was reacted with allylamine in acetonitrile at 50 °C for 5 h obtaining the N-allyl-1,1′-binaphthylazepine (S)-9 in 92% yield. The amine (S)-9 was deprotected by treatment with Pd(OAc)2 (2 mol%) in the presence of N,N-dimethylbarbituric acid (NDMBA) (0.3 equiv) and triphenylphosphine (10 mol%), leading to the 1,1′-binaphthylazepine (S)-10 in 54% yield. Finally, (S)-10 was bis-alkylated by the dibromide (S)-8 by refluxing in acetonitrile for 7 h in the presence of K2CO3, obtaining the ammonium salt (S,S)-6 in 74% yield. The biphenyl-binaphthylazepine salt (S)-7 was similarly prepared from 2,2′-bis(bromomethyl)-1,1′-biphenyl (11) (Scheme 3), in turn easily obtainable from commercially available 1,1′-diphenic acid. The dibromide 11 was converted into the hydroxylamine 12 by reaction with hydroxylamine hydrochloride at reflux in triethylamine for 2 h. The hydroxylamine 12 was then reduced in 96% yield to the corresponding biphenylamine 13 by treatment with zinc metal in the presence of a catalytic amount of indium (0.05 equiv) in a 1:1 mixture of EtOH/NH4Cl [13]. Finally, the ammonium salt (S)-7 was obtained in 62% yield by reaction of biphenylamine 13 with the dibromide (S)-8.
2.2 Asymmetric PTC Michael additions
The Michael addition of alkyl acetoacetates to enone 2 was performed at room temperature under nitrogen, in the presence of a base (K2CO3 or KOH) and a catalytic amount (10 mol%) of the chiral ammonium salt, affording the addition product 3. The latter was then hydrolyzed and decarboxylated by heating in water at 190 °C in a sealed tube, leading to the target compound trans-1. In Table 1 the results of the addition reaction using the Cinchona-derived salts 4 and 5 are reported.
Conjugate addition of ethyl acetoacetate to enone 2 in the presence of Cinchona-ammonium salts a
| Run | Acetoacetate (equiv) | Solvent | Base (equiv) | Time | Yield (%) b | ee (%) c,d (a.c.) e |
| 1 | 30 | – | K2CO3 (0.2) | 7 d | 60 | 68f (2S,3S) |
| 2g | 30 | – | K2CO3 (0.2) | 7 d | 60 | 64f (2R,3R) |
| 3h | 30 | – | K2CO3 (0.2) | 7 d | 0 | – |
| 4 | 3.0 | Toluene | K2CO3 (1.0) | 4 d | 53 | 47f (2S,3S) |
| 5 | 3.0 | Toluene | KOH (3.0) | 3 d | 48 | 28 (2S,3S) |
| 6h | 3.0 | Toluene | K2CO3 (1.0) | 4 d | 20 | – |
| 7 | 3.0 | THF | K2CO3 (1.0) | 6 d | 58 | 17 (2S,3S) |
| 8 | 3.0 | THF | KOH (1.0) | 3 d | 64 | 29 (2S,3S) |
| 9 | 3.0 | CH2Cl2 | K2CO3 (1.0) | 6 d | 50 | 25 (2S,3S) |
| 10 | 3.0 | CH2Cl2 | KOH (1.0) | 4 d | 50 | 16 (2S,3S) |
| 11 | 3.0 | Et2O | K2CO3 (1.0) | 2 d | 77 | 0 |
| 12 | 3.0 | Et2O | KOH (1.0) | 2 d | 49 | 27 (2S,3S) |
| 13i | 30 | – | K2CO3 (0.2) | 2 d | 50 | 74f (2S,3S) |
| 14i | 3.0 | Toluene | K2CO3 (1.0) | 2 d | 52 | 60f (2S,3S) |
a Reactions performed at RT, in the presence of salt 4 (0.1 equiv).
b Isolated yield in addition product 3.
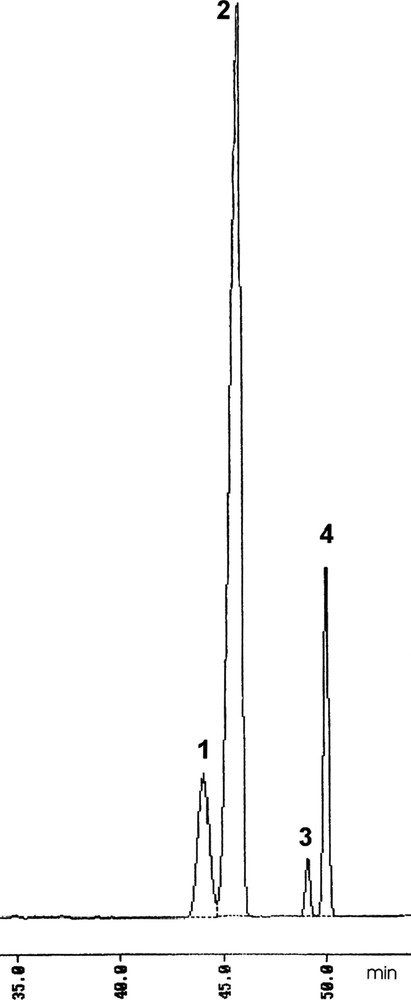
c In all the runs a 85:15 trans/cis ration for 1 was determined by GC.
d Ee of trans-1 determined by HPLC on Chiralcel OJ-H column.
e For assignment of absolute configuration, see [7].
f Ee of trans-1 determined by GC on a Cydex-B column.
g Catalyst 5 (0.1 equiv) was employed.
h No catalyst added.
i tert-Butyl acetoacetate was used.
By reacting enone 2 without solvent with an excess of ethyl acetoacetate, in the presence of K2CO3 (20 mol%) and salt 4 (10 mol%), product 3 was obtained in 60% yield, leading after decarboxylation to (2S,3S)-1 in 85:15 trans/cis diastereoselectivity and 68% ee 1. In the same conditions the pseudo-enantiomeric [14] catalyst 5 allowed to obtain the opposite enantiomer trans-(2R,3R)-1 with the same diastereoselectivity and slightly lower (64%) ee. Both reactions did not afford total conversion of the product and did not proceed further after 7 days. It has in fact to be taken into account that Cinchona-derived ammonium salts with a free hydroxyl function, like the present ones, can undergo rearrangements and decomposition when left for prolonged time in basic conditions [15]. Therefore, due to such decomposition, the amount of catalyst decreases progressively, then slowing down the reaction. Run 3 in fact reveals that, in these reaction conditions, the uncatalyzed process does not proceed at all. Chiral GC analysis of the reaction mixtures of 1 (Fig. 3) showed that in all the trials the same ee is obtained for both trans and cis stereoisomer and a 85:15 trans/cis ratio is constantly achieved, as obtained in the synthesis of racemic 1 via the acetoacetate addition to 2 in EtOH/EtONa [4,7]. We can then confidently consider the diastereomeric ratio simply due to the relative thermodynamic stability of the trans and cis isomer. It is in fact reasonable that the chiral salt just affects the facial addition of the acetoacetate anion to the prochiral C(3), but it does not exert any effect on the following enolate protonation which determines the absolute configuration at C(2). It must also be taken into account that in the analogous dihydrojasmonates the cis-isomer spontaneously isomerizes to the 90:10 trans/cis thermodynamic ratio outside the narrow 5–7 pH range [1,2c], therefore also compound 1 could be affected by such an equilibration. The conjugate addition of ethyl acetoacetate was slightly faster when performed in solution. In toluene and in the presence of catalyst 4 (run 4) 3 was recovered in 4 days, but lower (47%) ee of (2S,3S)-1 was obtained. The use of KOH in toluene (run 5) did not modify the yield in 3, but further lowered the ee to 28%. Interestingly, in toluene the reaction proceeds also without the catalyst (run 6) although with a very low conversion. Therefore it is reasonable that for prolonged reaction time, when catalyst decomposition can occur, the non enantioselective uncatalyzed reaction can compete, lowering the overall ee. As enantio-selectivity is concerned, an inverse behavior in respect to toluene was observed in THF (runs 7 and 8), were KOH afforded a 29% ee, higher than K2CO3, although still modest. Both in CH2Cl2 and Et2O (runs 9–12), with both K2CO3 and KOH, ee's lower than 30% were achieved. The reaction carried out in Et2O and in the presence of K2CO3 (run 11) instead afforded higher yields, but a racemic product. In order to verify if an increase of the size of the alcoholic moiety of the acetoacetate could enhance the enantio-selectivity we tested tert-butyl acetoacetate in this reaction, using the best reaction conditions (K2CO3 either without solvent or in toluene) established. The use of the tert-butyl ester allowed a faster reaction in respect to the ethyl one and higher ee's. In fact, a 74% ee of (2S,3S)-1 was obtained when the reaction was carried out without solvent (run 13) and a 60% ee was achieved in toluene (run 14) 2. The higher enantio-selectivity obtained when tert-butyl acetoacetate was used can be ascribed either to its larger size, which ensure a better facial discrimination, or to the slightly higher lipophilicity of its anion, which accelerates the reaction. In fact, as showed before, when the reaction is too slow a decrease of ee could occur. The 1,1′-binaphthylazepine salts (S,S)-6 and (S)-7 were then tested, still using the optimized reaction conditions (Table 2). With both (S,S)-6 and (S)-7 the (2R,3R) of trans-1 was obtained. Unfortunately, with both catalysts the addition reaction was rather slow and, most importantly, low enantio-selectivity (ee < 12%) was obtained both in toluene and in absence of solvent. Probably these salts are too lipophilic to act, in our reaction conditions, as efficient carrier of the very polar acetoacetate anion.
Conjugate addition of ethyl acetoacetate to enone 2 in the presence of 1,1′-binaphthylazepine ammonium salts a
| Run | Catalyst | Acetoacetate (equiv) | Solvent | Base (equiv) | Time (days) | Yield (%) b | ee (%) c,d (a.c.) e |
| 1 | (S,S)-6 | 30 | – | K2CO3 (0.2) | 6 | 50 | 10 (2R,3R) |
| 2 | (S,S)-6 | 3.0 | Toluene | K2CO3 (1.0) | 6 | 47 | 9 (2R,3R) |
| 3 | (S)-7 | 30 | – | K2CO3 (0.2) | 6 | 60 | 12 (2R,3R) |
| 4 | (S)-7 | 3.0 | Toluene | K2CO3 (1.0) | 6 | 40 | 8f (2R,3R) |
a Reactions performed at RT, in the presence of 10 mol% of catalyst.
b Isolated yield in addition product 3.
c In all the runs a 85:15 trans/cis ration for 1 was determined by GC.
d Ee of trans-1 determined by HPLC on Chiralcel OJ-H column.
e For assignment of absolute configuration see Ref. [7].
f Ee of trans-1 determined by GC on a Cydex-B column.
3 Conclusions
In this investigation a wide screening of the reaction condition for the stereoselective conjugate addition of acetoacetates to enone 2 under PTC conditions have been performed, establishing an optimized procedure which allowed to obtain trans isomer of the fragrance magnolione (1) in 85:15 trans/cis ratio and ee up to 74%. The use of salts 4 and 5, derived from pseudo-enantiomeric alkaloids, allowed to obtain both enantiomers of trans-1 with comparable enantio- and diastereoselectivity. Lower ee's were instead obtained using the less polar 1,1′-binaphthylazepine salts (S,S)-6 and (S)-7. Even if the enantioselectivities obtained are still moderate this approach, carried out under mild conditions, appears very promising.
This investigation represents in fact an important step in order to undertake the study of the structure/odor relationship of such artificial fragrance, as well as to make available to the perfume industry other valuable ingredients.
4 Experimental section
4.1 General procedures
Melting points were determined with a Kofler hot-stage apparatus and are uncorrected. 1H NMR and 13C NMR spectra were recorded in CDCl3 either on a Varian Inova spectrometer or on a Bruker Aspect 300 spectrometer. Optical rotations were measured by a JASCO DIP-370 digital polarimeter. Enantiomeric excess of compound 1 was determined either or by HPLC on Chiralcel OJ-H c.s.p. (hexane/i-PrOH = 99:1 v/v; flow = 0.5 ml/min; λ = 280 nm) or by GC analysis on a Cydex-B c.s.p. 2-Pentyl-2-cyclopenten-1-one (2) and ethyl acetoacetate were distilled prior their use. KOH and K2CO3 were pulverized and dried under vacuum. THF, Et2O, and toluene were freshly distilled prior the use on sodium benzophenone ketyl and stored under nitrogen atmosphere. CH2Cl2 and CH3CN were freshly distilled on CaH2 and stored under nitrogen atmosphere. Triethylamine was distilled over CaH2 and stored under nitrogen on KOH. Enantiopure N-9-anthracenylmethyl quininium chloride (4) and N-9-anthracenylmethyl quinidinium chloride (5) were prepared according to literature procedures [2d,8]. Enantiopure (S)-2,2′-bis(bromomethyl)-1,1′-binaphthalene (S)-8 was prepared as previously described [9a]. Chiral ammonium salt (S,S)-(–)-6 was prepared as reported by Ooi et al. [11] starting from dibromide (S)-8. 2,2′-Bis(bromomethyl)-1,1′-biphenyl (11) was prepared by PBr3 bromination of 2,2′-bis(hydroxymethyl)-1,1′-biphenyl, in turn obtained by LiAlH4 reduction of commercially available diphenic acid. Analytical TLC were performed on 0.2 mm silica gel plates Merck 60 F-254 and column chromatographies were carried out with silica gel Merck 60 (70–230 mesh). GC analyses were carried out on GC/MS Hewlett Packard 5080 series II, detector HP 5971, column Supelco 57300-U (polydimethylsiloxane phase, PDMS). Chiral GC analyses were carried out on a Perkin Elmer Autosystem gas chromatograph (detector: FID; injector mode: split; carrier gas: He) equipped with an SGE Cydex-B capillary colum (25 m × 0.22 mm ID, 0.25 μm film).
4.2 6,7-Dihydro-5H-dibenz[c,e]azepine-N-hydroxy (12)
A solution of dibromide (11) (6.0 g, 18.0 mmol), triethylamine (53 ml) and hydroxylamine hydrochloride (3.7 g, 54.0 mmol), was heated at reflux and stirred for 2 h under nitrogen atmosphere. The mixture was then filtered under vacuum and the resulting solution was distilled to remove the triethylamine. The crude product was purified by column chromatography on silica gel (from petroleum ether/diethyl ether 4:1 v/v, to petroleum ether/diethyl ether 2:1 v/v) gave product 12 (1.7 g, 50% yield).
1H NMR (300 MHz, CDCl3) δ (ppm): 3.15 (d, J = 12 Hz, 2H), 3.95 (d, J = 12 Hz, 2H), 7.5 (m, 9H); 13C NMR (75 MHz, CDCl3) δ (ppm): 60.44, 127.81, 129.51, 130.15, 133.92, 14.95.
4.3 6,7-Dihydro-5H-dibenz[c,e]azepine (13)
The hydroxylamine 12 (1.7 g, 8.0 mmol) was dissolved into a 1:1 solution of EtOH and saturated aqueous NH4Cl (40 ml; pH 6). Indium powder (5 mol%, 0.046 g, 0.4 mmol) and zinc powder (2 equiv, 1.04 g, 16.0 mmol) were added and the mixture was heated at reflux for 7 h. The mixture was cooled, filtered over Celite and concentrated. A solution of saturated aqueous Na2CO3 was then added and the mixture was extracted with ethyl acetate. The organic phase was dried over anhydrous Na2SO4 and concentrated to afford 1.5 g (95% yield) of 13 as a white solid.
M.p. = 229–231 °C; 1H NMR (500 MHz, CDCl3) δ (ppm): 4.04 (s, 5H), 7.48 (m, 2H), 7.57 (m, 4H), 7.64 (d, J = 7 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ (ppm): 45.90, 128.61, 129.22, 129.54, 130.50, 131.27, 140.81.
4.4 Chiral ammonium salt (S)-(–)-7
A mixture of 13 (0.186 g, 0.95 mmol), dibromide (S)-8 (0.467 g, 1.1 mmol), and K2CO3 (0.204 g, 1.5 mmol) in CH3CN was heated at reflux, and stirred for 48 h. The resulting mixture was poured into water and extracted with CH2Cl2. The organic extracts were dried over anhydrous Na2SO4 and concentrated. The residue was purified by column chromatography on silica gel (MeOH/CH2Cl2 1:9 v/v) to furnish 0.376 g (62% yield) of (S)-7 as a white solid.
[α]D21 = –94 (c = 1.1, CHCl3); 1H NMR (500 MHz, CDCl3) δ (ppm): 3.97 (d, J = 13 Hz, 2H), 4.06 (d, J = 13 Hz, 2H), 4.35 (d, J = 13 Hz, 2H), 4.80 (d, J = 13 Hz, 2H), 7.40 (t, J = 8 Hz, 2H), 7.46 (d, J = 9 Hz, 2H), 7.64 (t, J = 7 Hz, 4H), 7.76 (m, 4H), 7.89(d, J = 6 Hz, 2H), 8.08 (dd, J = 8, 9 Hz, 4H), 8.30 (d, J = 9 Hz, 2H); 13C NMR (125 MHz) (CDCl3) δ (ppm): 61.0, 62.0, 125.9, 126.7, 127.8, 127.9, 128.0, 128.1, 129.0, 129.8, 130.1, 131.2, 131.8, 132.3, 132.4, 134.8, 137.1, 141.3.
4.5 2-Pentyl-3-(1-carbethoxy-2-oxopropyl)-1-cyclopentanone (3)
4.5.1 General procedure without solvent
To a solution of 2 (203 mg, 1.3 mmol) in the alkyl acetoacetate (39.0 mmol, 30 equiv), the catalyst (0.11 equiv), and potassium carbonate (54 mg, 0.392 mmol, 0.028 equiv) were added in sequence. After stirring at room temperature for some days (see Tables 1 and 2), the reaction mixture was diluted with diethyl ether (40 ml). The organic layer was washed in sequence with 10% aqueous HCl (2 × 10 ml), water (10 ml) and brine (2 × 10 ml). The organic layer was then dried over anhydrous Na2SO4, filtered and evaporated. The crude pale yellow oil was distilled under reduced pressure to remove the alkyl acetoacetate. Finally, chromatography on silica gel (petroleum ether/diethyl ether 70:30 v/v) of the crude residue yielded product 3 as a colorless oil. Product 3 was obtained, by addition of ethyl acetoacetate to 2, as a mixture of four diastereoisomers. Here only the 1H and 13C NMR spectra of the two more abundant isomers have been accounted.
1H NMR (600 MHz, CDCl3): δ 0.87 (t, J = 7 Hz, 3H); 1.23 (m, 5H); 1.30 (t, J = 7 Hz, 3H); 1.40–1.45 (m, 2H); 1.57–1.70 (m, 2H); 1.96 (m, 1H); 2.16 (m, 2H); 2.27 (s, 3H); 2.33 (m, 1H); 2.65–2.73 (m, 1H); 3.47–3.57 (d, J = 7 Hz, 1H); 4.22 (q, J = 7 Hz, 2H). 13C NMR (150.9 MHz, CDCl3) Isomer 1 δ 13.97 (CH3); 14.09 (CH3); 22.41 (CH2); 23.91 (CH2); 26.04 (CH2); 26.04 (CH2); 28.43 (CH2); 29.60 (COCH3); 31.93 (CH2); 37.18 (CH2); 39.80 (CH); 52.24 (CH); 61.50 (COCH2); 62.18 (CH); 168.55 (quat, COO); 201.88 (quat, CO); 219.14 (quat, CO). Isomer 2 δ 13.96 (CH3); 14.03 (CH3); 22.39 (CH2); 24.95 (CH2); 24.95 (CH2); 25.89 (CH2); 28.78 (CH2); 29.56 (COCH3); 32.00 (CH2); 37.03 (CH2); 39.72 (CH); 52.51 (CH); 61.66 (COCH2); 63.84 (CH); 168.70 (quat, COO); 201.88 (quat, CO); 219.02 (quat, CO). MS (EI): m/z 282 (M+, 1), 239 (1), 212 (1), 169 (62), 153 (79), 139 (67), 131 (100), 123 (19), 97 (25), 83 (43), 55 (27), 43 (72). Anal. Calc. for C16H26O4: C, 68.06; H, 9.28. Found: C, 68.0; H, 9.1.
4.5.2 General procedure with solvent
A solution of enone 2 (1.0 equiv), catalyst (0.11 equiv), base (1.0 equiv) and alkyl acetoacetate (3.0 equiv) in a dry solvent (6.5 ml) was stirred at room temperature for several days (see Tables 1 and 2). The reaction mixture was diluted with diethyl ether and the organic layer was washed in sequence with 10% aqueous HCl (2 × 10 ml), water (10 ml) and brine (2 × 10 ml). The organic phase was dried over anhydrous Na2SO4, filtered and evaporated. The crude product, purified by chromatography on silica gel (petroleum ether/diethyl ether 70:30 v/v), afforded 3 as a colorless oil.
4.6 2-Pentyl-3-(2-oxopropyl)-1-cyclopentanone (Magnolione) (1)
A mixture of 3 (1.5 mmol) in water (750 μl) was introduced in a 2 ml vial. The vial was sealed by flame and the mixture was heated at 190 °C for 12 h. The cooled mixture was extracted with diethyl ether (10 ml) and the organic phase was dried over anhydrous Na2SO4, then filtered and evaporated. The crude residue, purified on silica gel (petroleum ether/diethyl ether 60:40), afforded 1 as a light yellow oil. GC analysis showed an 85/15 trans-1:cis-1 ratio. The use of catalyst 4 gave (2S,3S)-1 as major enantiomer, the opposite was obtained when catalysts 5 (S,S)-6 and (S)-7 were used.
For trans-1: 1H NMR (600 MHz, CDCl3): δ 0.88 (t, J = 7 Hz, 3H); 1.25 (m, 2H); 1.29 (m, 3H); 1.38 (m, 2H); 1.53 (m, 2H); 1.74 (m, J = 10 Hz, J = 6 Hz, J = 2 Hz, 1H); 2.13 (m, J = 9 Hz, J = 11 Hz, J = 19 Hz, 1H); 2.19 (s, 3H); 2.25 (m, 1H); 2.32 (m, 1H); 2.35 (m, 1H); 2.46 (dd, J = 9 Hz, J = 17 Hz, 1H); 2.75 (dd, J = 4 Hz, J = 17 Hz, 1H); 13C NMR (150.9 MHz, CDCl3): δ 13.85 (CH3); 22.30 (CH2); 26.26 (CH2); 27.24 (CH2); 27.77 (CH2); 30.45 (COCH3); 36.86 (CH); 37.72 (CH2); 47.82 (COCH2); 54.12 (CH); 207.65 (quat, CO); 220.09 (quat, CO). MS (EI): m/z 210 (M+, 4), 153 (52), 140 (21), 125 (12), 97 (18), 82 (100), 55 (18), 43 (49). Anal. Calc. for C13H22O2: C, 74.24; H, 10.54. Found: C, 74.21; H, 10.23.
Acknowledgements
Financial support from MIUR-COFIN2002 ‘Synthesis of flavors and fragrances’ and Università della Basilicata is gratefully acknowledged.
1 See Ref. [7] for assignment of the relative and absolute stereochemistry of the major stereoisomer of 1 by NMR and CD spectroscopy, respectively.
2 It is noteworthy that in the addition product 3, coming from reaction of tert-butyl acetoacetate, a better chromatographic separation of the cis and trans isomers is observed. Therefore, collecting different chromatographic fractions, enriched samples of both trans-1 and cis-1 were obtained.