

1 Introduction
The adaptor protein Grb2, encompassing one SH2 and two SH3 domains, is an important element in Ras signaling. Its SH2 domain interacts at the level of tyrosine phosphorylated sequences with proteins such as members of the erbB family, and its SH3 domains bind proline-rich sequences of proteins like Sos, the exchange factor of Ras [1]. Upon growth-factor stimulation of RTK, the Grb2-Sos complex is recruited to the membrane and can promote Ras-GTP binding and the subsequent activation of the mitogen activating protein (MAP) kinase cascade, involved in cell growth and differentiation [2]. Anarchic cell proliferation, observed in some leukemia and in breast and ovarian cancers has been related to dysfunctioning of cytoplasmic proteins or receptors with tyrosine kinase activity (PTK or RTK) coupled to p21 Ras activation [3]. Thus, Grb2 constitutes an attractive target for the design of new anti-tumor therapeutic agents. To provide such antiproliferative agents, synthetic phosphotyrosine-containing peptides were therefore designed that inhibit the interaction between the SH2 domain of Grb2 and either RTKs or adaptators like Shc with IC50 values in the 10–8–10–9 M range [4]. Other approaches, which consist into inhibiting the interaction between Sos and the SH3 domains of Grb2 were also developed [4a,5]. Thus, with the aim of attaining simultaneous interactions with both SH3 domains of Grb2, peptide dimers have been designed in our laboratory by coupling two proline-rich sequences from Sos using linkers of different sizes.
We describe here our approaches developed in order to inhibit Grb2 interactions either at the level of SH3 domains or SH2 domain.
2 Inhibitors of Grb2 SH3 domains
2.1 Conception and biological data
To inhibit Grb2-Sos interaction we took advantage of the presence of two spatially close SH3 domains in Grb2 observed in its crystallized structure [6]. We then used the 3D structures of each SH3 domain complexed with a proline-rich peptide resolved by NMR [7] to design proline-rich peptide dimers which could interact with both SH3 domains. When they can bind twice as monomers do, dimers are expected to have a higher affinity for their target. Using molecular modeling, two proline-rich peptides were docked to each SH3 domain according to the geometry determined by NMR. Carboxy terminals of both peptides appear adjacent one to another, thus, we linked them by a connector containing two amino groups, which was optimized as a lysine [8].
The monomer (VPPPVPPRRR) which was selected is characterized by low affinity for Grb2 (Kd around 18 μM). The dimers, constituted by two sequences linked by their terminal carboxyl groups with the amino groups of an appropriate linker, showed affinity for Grb2 in the range of 40 nM, which constitutes a more than 400-fold increase. These dimers were called ‘peptidimers’. A peptidimer with a lysine linker was shown to inhibit Grb2-Sos association on ER22 cells homogenate (fibroblasts overexpressing EGFR) stimulated with EGF. The Grb2-Sos inhibition was complete with 5 μM of the monomer while 50 nM of the dimer was sufficient. The specificity of the peptidimer was also shown since it has no affinity for the proteins PI3K, Nck, Crk and Mona, which encompass one or more SH3 domains [8].
Although the peptidimers were quite efficient to inhibit Grb2-Sos interaction on a cellular homogenate, they did not show cellular effects by themselves. Thus, they were subsequently coupled to a vector molecule that allows cell diffusion. Penetratin, a peptide derived from the homeodomain of Antennapedia, was chosen [9]. On tumor model NIH3T3 cells transfected with the oncogenic tyrosine kinase receptor HER2, the peptidimer-conjugate is able to inhibit the formation of colonies in a plating efficiency test with an ED50 value around 0.05 μM (Fig. 1).

Effect of vectorized peptidimer on NIH3T3/HER2 cells on a plating efficiency test.
We then took advantage of the design by Nguyen et al. of a peptoid with high affinity for the N-SH3 domain of Grb2 [5]. In this peptoid, a proline residue (in bold in the sequence (VPPPVPPRRR) was replaced by a N-alkylated glycine, N-(S)-(α-phenylethyl)glycine (peG), providing additional interactions with the SH3 domain (Fig. 2). We have synthesized corresponding peptoid analog dimers which exhibit so high affinities for Grb2 that their measure by classical fluorescence was not possible (Table 1).

Conception of N-alkylated peptoids.
Affinities (expressed as Kd values measured by fluorescence, in μM) and inhibitory constant (Ki in μM expressed as displacement of Grb2/VPPPVPPRRR interaction). PeG: (S)-(α-phenyl)ethylglycine; N.M.: not measurable
Thus, we have therefore developed a test to quantify such affinities. Grb2 was pulled-down from cellular extract by the VPPPVPPRRR peptide bound on CNBr activated sepharose beads, with or without the competitor. This allowed us to access to Ki values. In this test, peptoid dimers exhibited Ki with subnanomolar value [10] (Table 1). These compounds are not active on cells and the vectorized molecules are now under preparation to evaluate their potential cellular activity.
2.2 Synthesis of the dimers
Peptide synthesis (Fig. 3) was carried out on solid phase with Fmoc (9-fluorenylmethoxycarbonyl) chemistry.
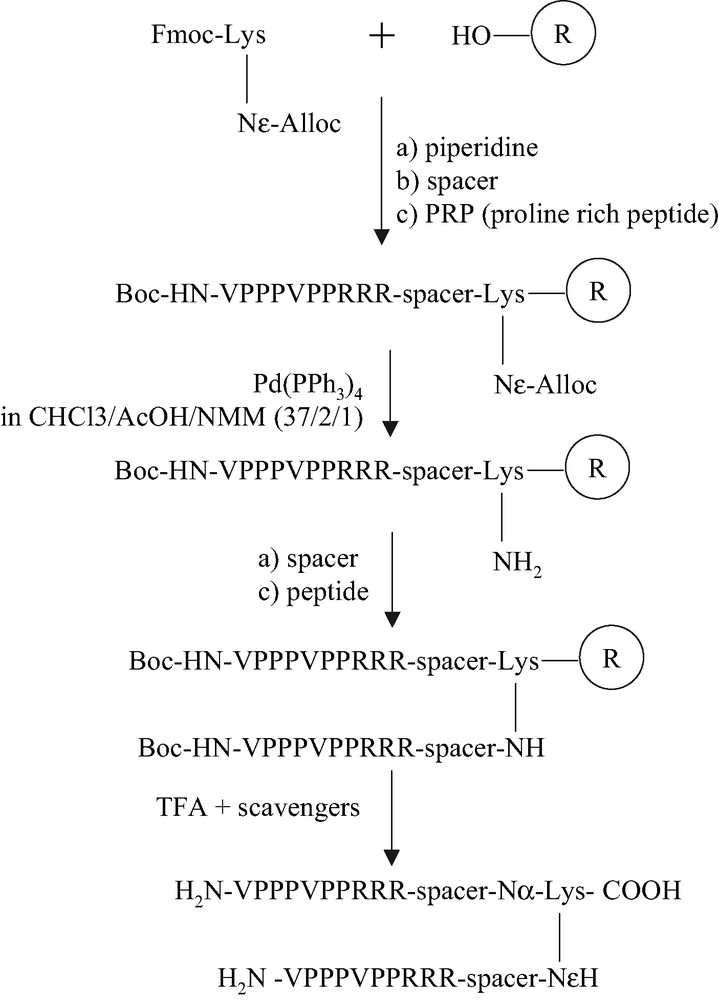
Synthesis of peptidimers.
For the symmetric peptide dimers, the di-Fmoc protected linker was loaded on an HMP resin. After deprotection of both Fmoc groups of the linker residue by a solution of 20% piperidine, residues of spacers (6-aminohexanoic acid, Aha) and both the proline-rich sequences were extended simultaneously. Concerning the asymmetric peptide dimers, Fmoc-Lys(Alloc)-OH was used in the place of Fmoc-Lys(Fmoc)-OH (Fig. 3). After deprotection of the Fmoc group and introduction of the first peptide sequence, with the last N-terminal residue in Boc form on the α-NH2 group of the last valine residue, the Alloc protecting group of the side chain ɛ-NH2 of lysine was removed by 3 equiv of Pd(PPh3)4 in a mixture of CHCl3/AcOH/N-methylmorpholine (37:2:1) for 2 h under nitrogen at room temperature [10]. The second proline-rich sequence was then introduced on the side chain of the lysine. For the synthesis of N-alkylated peptide dimers, instead of coupling a proline, bromoacetic acid was added by double-coupling. The bromides were then substituted by (S)-(α-phenyl)ethylamine in DMSO. The valine residues were coupled by double-coupling with PyBop/DIEA in DMF. The remaining residues were introduced by the DCC/HOBt coupling (Fig. 4). Peptides were then purified by reverse phase HPLC after deprotection and cleavage from the resin. The molecular weight of peptides were verified by ion electrospray mass spectrometry.

Synthesis of peptoid dimers.
3 Inhibitors of Grb2 SH2 domain
3.1 Conception and affinity of the inhibitors for Grb2-SH2 domain
The SH2 domains are highly folded structures containing one central beta-sheet crossing at the surface of the domain with two helices on each side. A recognition groove for phosphopeptides appears on the surface of the domain, where the pTyr (pY) side chain can interact with basic residues of the groove and the aminoacid C-terminal to pY can interact with other residues providing the specificity of the recognition [11]. Rahuel et al. [12] have resolved by X-ray crystallography the structure of Grb2 SH2 complexed with phosphopeptide KPFpYVNV, and observed that the complexed peptide adopts a β-turn conformation due to the presence of a tryptophan residue in the groove that causes a large steric hindrance. Two aminoacid C-terminal to pY, especially the Asn residue, appeared essential for Grb2 SH2 domain recognition. Based on the structural data, different series of N- or C-terminal modified compounds, including the pY-X-N sequence with X as hydrophobic aminoacid have been designed in order to obtain small molecules. In our case, we exploited the fact that the Shc 239/240 sequence contains the consensus motif pYXN in which X is replaced by a tyrosine or phosphotyrosine residue [13]. Accordingly, compounds with the sequence pY-pY-N, ending with mAz (meta-aminobenzyloxycarbonyl) at the N-terminal part [14] and by a NH2 group at the C-terminal part to allow formation of a beta-turn, were designed. The series was further optimized to increase the affinity for the Grb2 SH2 domain (Table 2).
Affinities (expressed as Kd values in nM) of different molecules, showing the importance of both the phosphorylation and the alpha methylation on the second tyrosine residue
| Molecule | Kd (nM) |
| mAZ-pY-Y-N-NH2 | 150 |
| mAZ-pY-pY-N-NH2 | 6 |
| mAZ-pY-(αMe)pY-N-NH2 | 3 |
| mAZ-pY-(αMe)Y-N-NH2 | 250 |
| mAZ-pY-(αMe)(4-CH2COOH)F-N-NH2 | 60 |
| mAZ-pY-(αMe)(4-COOH)F-N-NH2 | 45 |
| mAZ-pY-(D,L)(αMe)(4-CH2PO3H2)F-N-NH2 | 70 |
| mAZ-pY-(αMe)(4-PO3H2)F-N-NH2 | 5 |
And to promote the β-turn structure, an α-methyl substitution was included on the second pY. The affinity of the phosphopeptides was measured through a fluorescent test and compared to that obtained by Garcia-Echeverria et al. [14] for mAz-pY-Ac6c-N-NH2 (Ac6c: 1-aminocyclohexanecarboxylic acid). As expected, the phosphorylation of both Tyr is essential. The phosphopeptide mAZ-pY-(αMe)pY-N-NH2 exhibits one of the highest affinities for the SH2 domain of Grb2 reported so far. In order to explain its important increased affinity, the peptide was docked on the surface of the SH2 domain. Reduction in peptide flexibility appears to provide a favorable orientation for additional stabilizing interactions between the phosphate group of the (αMe)pTyr and the Asn 143 and Arg 142 residues. This was confirmed by X-ray of Grb2-SH2 domain crystallized with the phosphopeptide [16].
Since cellular phosphatases are likely to hydrolyze the phosphate group of phosphotyrosine that is necessary for Grb2 affinity, we have replaced in our series of molecules the phosphate group of the (αMe)pY, by phosphatases resistant carboxylate or phosphonate substituents [15,17]. The carboxylate or methylphosphonate analogs showed lower affinities than the peptide mAZ-pY-(αMe)pY-N-NH2 (Kd = 3 nM), which is likely related to the presence of only one negative charge on the carboxylate group or the decreased acidity of methylphosphonate group that could reduce the ionic interactions with the Asn 143 and Arg 142 side chains of the SH2 domain. However, a mimetic with phosphonate directly attached to the aromatic ring, which having lower pKa, in the peptide sequence has similar Kd value as that of the (αMe)pY containing peptide.
3.2 Design and test of prodrugs
Although phosphate or phosphonate peptides show high affinity for SH2 domains, they cannot enter cells easily. This problem can be solved by conjugating the molecule with a vector peptide [9,18], or by derivating them to prodrugs [19]. Compound mAZ-pY-(αMe)pY-N-NH2 was chosen in order to prepare prodrugs, in which the phosphate groups were protected with S-acetyl thioethyl ester (SATE ou MeSATE).
This modification has been used by the group of Imbach to deliver a HIV antiviral drug into cells [20], and we successfully used it to deliver phosphopeptides derived from Shc pY317 (Ac-PFpY(SATE2)VNVP-NH2) into tumor cells [21]. The prodrugs thus obtained are very hydrophobic and can easily enter cells, where they are degraded by esterases.
As expected, the doubly-modified prodrug (named di-SATE prodrug), mAZ-pY(SATE)2-(αMe)pY(SATE)2-N-NH2, has no in vitro affinity for the SH2 domain of Grb2 in this prodrug form. Nevertheless, it is able, at very low dose (ED50 = 0.1 μM), to inhibit the growth of NIH3T3 cells transfected with HER2 in colonies on soft agar.
3.3 Synthesis of the compounds
For the synthesis of (αMe)pTyr mimetics, the two carboxylate (αMe)pTyr mimetics were prepared enantioselectively by the method of Williams and Im [22]. The phosphonates were obtained in racemic forms by phase-transferred catalytic alkylation of the benzylidene of alaninate [23]. The suitably protected mimetic aminoacids were then incorporated into the peptidic sequence by solid-phase peptide synthesis, using HATU/HOAt as coupling agents since the mimetics are hindered by their α-methyl group [15,17].
The prodrug mAZ-pTyr(SATE2)-(αMe)pTyr(SATE2)-Asn-NH2 was obtained according to the Fig. 5.
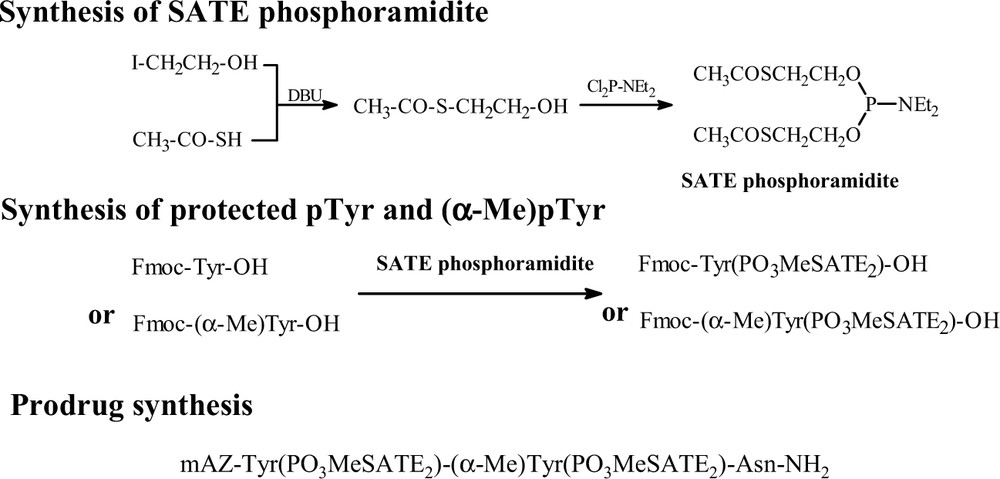
Principle of prodrug synthesis.
For the synthesis of the prodrug, we prepared at first the SATE phosphoramidite, which was then used in the phosphorylation of suitable protected Tyr or (αMe)Tyr. The SATE protected pTyr and (αMe)pTyr were coupled on solid phase or solution phase peptide synthesis to obtain the final molecule (Fig. 5).
4 Conclusion
To conclude this review, we have obtained peptidimers and peptoid dimers targeting SH3 domains of Grb2 that show very high affinities for Grb2. Conjugated with penetratin, the peptidimer can enter cells to inhibit Grb2/Sos interaction and MAPK phosphorylation. It displays an antiproliferative effect on tumor cells involving the Ras signaling pathway in response to tyrosine kinase protein activation. Moreover, it does not seem to develop toxicity in systems which do not overexpress the Ras signaling pathway. On SH2 domain, it was possible to optimize the phosphorylated peptides and we obtained modified peptides with very high affinity for Grb2 in vitro whose prodrug derivatives are currently under investigations in vivo.
Acknowledgments
We acknowledge the ‘Ligue Nationale contre le Cancer, Comité de Paris’ for financial support.