

1 Introduction
Processes in which a silicon group attached to a carbon framework is used to control the stereochemistry have now become very common in organic synthesis [1]. A stereogenic center bearing a silicon substituent has thus been shown to transfer its chirality to the neighboring prostereogenic center with generally high efficiency in many organic processes. Such a concept has been amply verified in ionic (electrophilic and nucleophilic) and pericyclic processes. Reliable models have been developed which allow the prediction of the stereochemical outcome of electrophilic functionalization of chiral allylsilanes and β-silyl enolates (Fig. 1) [1c]. 1,2-Diastereocontrol in reactions of allylsilanes is usually explained invoking an approach of the electrophile (i.e. X–Y) anti relative to the silicon group, in a conformation such as Ia (Fig. 1) where the smallest group (e.g. H) eclipses the double bond (inside position), the silicon–carbon bond being thus perpendicular to the olefin, allowing the stabilization of the incipient carbocation (σ–π stabilization) [2]. The large size of the silicon group relative to the two other groups usually favors one conformation relative to the others. This is particularly true with Z-olefins where important A1,3 strain is involved [3]. Conformation Ib having the medium-sized group RM inside experiences strong A1,3 interactions between RM and RZ, thus explaining the generally higher diastereocontrol observed with (Z)-allylsilanes as compared to (E)-allylsilanes [1]. It is worthy of note that the transition state conformation Ia closely resembles that of the allylsilane in the ground state, in which interactions between the σC–Si bond and the olefin raise the HOMO of the allylsilane, making it more reactive towards electrophiles [4]. Calculations of electrostatic potentials corresponding to the diastereotopic faces also led to the conclusion that the face opposite to silicon was more reactive towards electrophiles. Therefore, it appears that electronic and steric effects reinforce each other in chiral allylsilanes, both contributing to the high level of stereocontrol generally observed in their reactions with electrophiles.
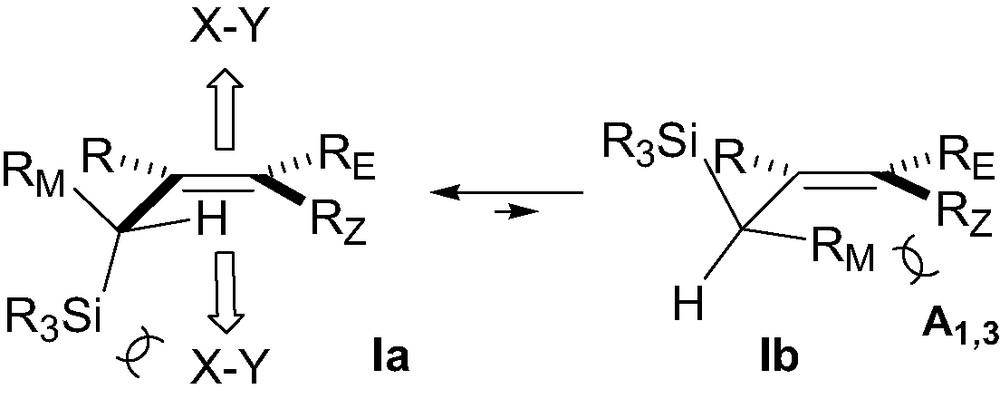
Transition-state model for electrophilic functionalization of allylic type chiral organosilicon derivatives.
In cyclic systems, locked conformations provide sufficient steric differentiation to lead to high anti-stereocontrol, whatever the nature of the silicon group. A similar treatment may be applied to nucleophilic addition to α-silylcarbonyl compounds, the nucleophilic species approaching anti relative to the bulky silicon group on the side of the smallest group (i.e. H), in a Felkin-Anh type conformation II (Fig. 2) [5].
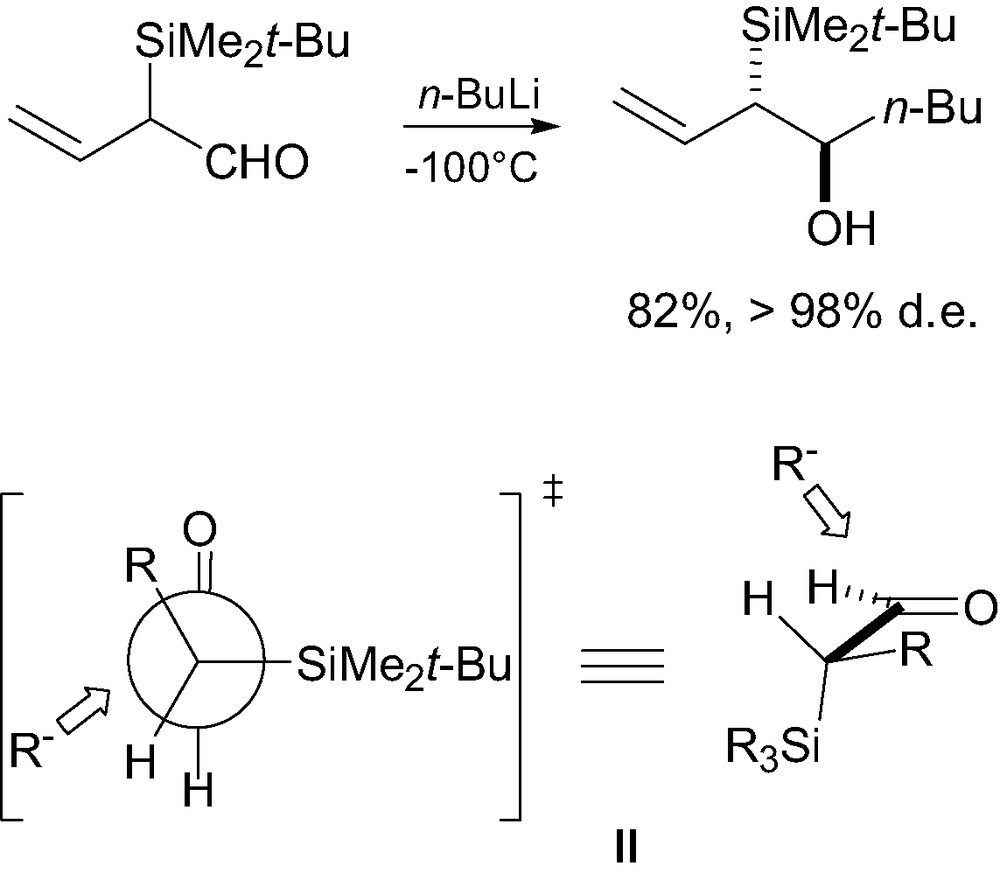
Transition state model for nucleophilic functionalization of α-silylcarbonyl compounds.
In contrast, very few studies have been carried out on the 1,2-stereocontrol arising from reactions on a carboradical center adjacent to a stereogenic center carrying a silicon group. Such β-silylcarboradicals are weakly stabilized (between 2 and 4 kcal/mol) as compared to their non-silylated radical counterpart, due to hyperconjugation (σ–π) or homoconjugation ((p–d)π) [6] and are known to react with various radical traps. It is only recently that several investigations have been reported, showing that useful level of 1,2-stereoinduction could be obtained. We describe in this context some of our most recent findings in this area, along with relevant studies from others, which should provide a broader view of the progress on the stereocontrol in reactions involving chiral β-silyl carboradicals (Fig. 3). This account will focus on two main topics, including acyclic 1,2-stereoinduction (i.e. 1 → 2, Fig. 3) and stereocontrol (1.2 and 1.5) arising during 5-exo-trig cyclizations of hexenyl radicals of type 3.
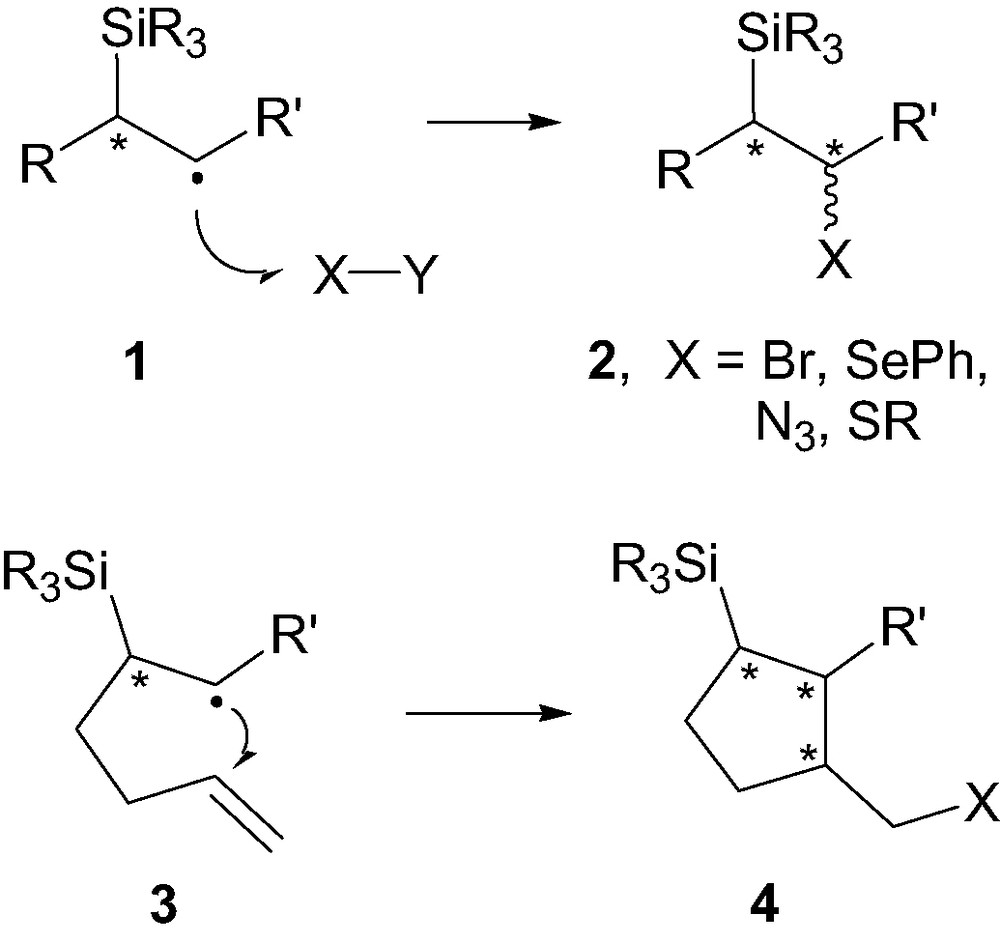
1,2-Stereoinduction in cyclic and acyclic free-radical processes.
2 Stereocontrol in reactions of acyclic β-silyl radicals
2.1 Allylic strain model
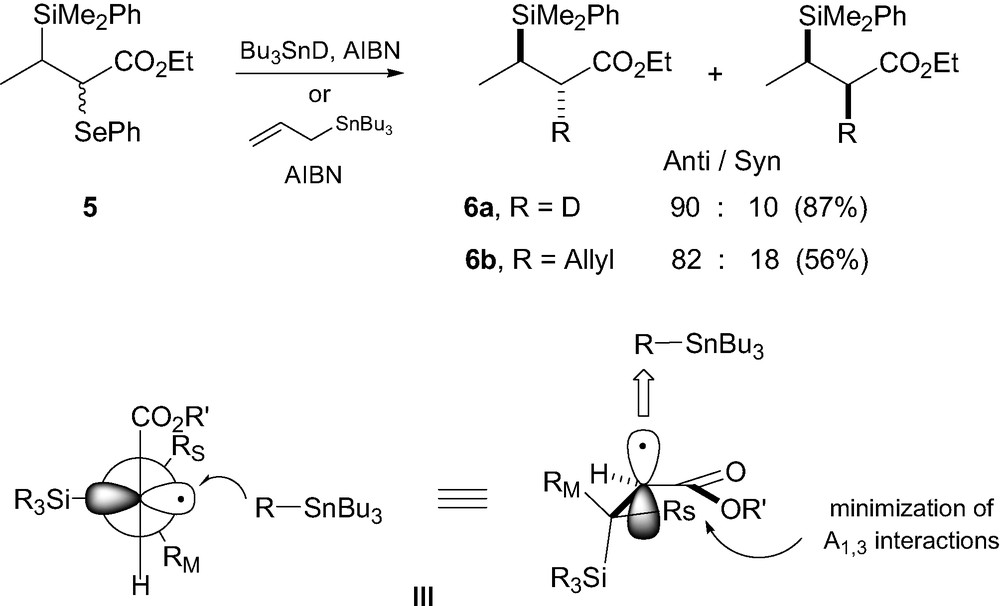
The first systematic study regarding stereocontrolled reactions on acyclic β-silyl carboradicals precursors has been carried out by Hart and Krishnamurthy [7]. They showed that deuteration and allylation of radical precursors 5 led to the corresponding anti-deuterated and allylated products 6a–b as major compounds with reasonable level of stereocontrol, comparable to those obtained using ionic processes (Scheme 1). In order to explain their results, they proposed a transition state model III based on allylic strain [3,8]. Such radical intermediates are assimilated to allylic systems due to the conjugation of the radical with the ester group. A radical adjacent to a carbonyl group thus behaves as a planar heteroallyl radical system (a radical enolate) which experiences A-1,3 strain [9]. Assuming that the smallest substituent RS (i.e. H) occupies the ‘inside’ position to minimize A-1,3 strain, the reagent should approach anti relative to the largest substituent RL (i.e. SiR3) to minimize torsional strain in the transition state [10].
A similar radical process, controlled by the allylic strain has been reported more recently by Miura et al. [11]. Allylsilane 8 [12] was thus found to add to α,β-unsaturated esters 7 and alkynes to provide allylsilylation products with generally good yields (Scheme 2). With olefins, the reaction is believed to proceed through the addition of the tris(trimethylsilyl)silyl ((Me3Si)3Si) radical to the olefin to generate a β-silyl carboradical which has enough lifetime to add intermolecularly to 8 to provide the addition product 9 and regenerate the silyl radical (Me3Si)3Si. The formation of the major anti-diastereomer 9 was rationalized invoking transition state IVa, closely related to the model III above (Scheme 1). Kopping et al. [13] observed similar trends for hydrosilylation of olefins 10 which occurred with good to excellent levels of stereocontrol, leading to the syn product 11 presumably through transition state IVb.
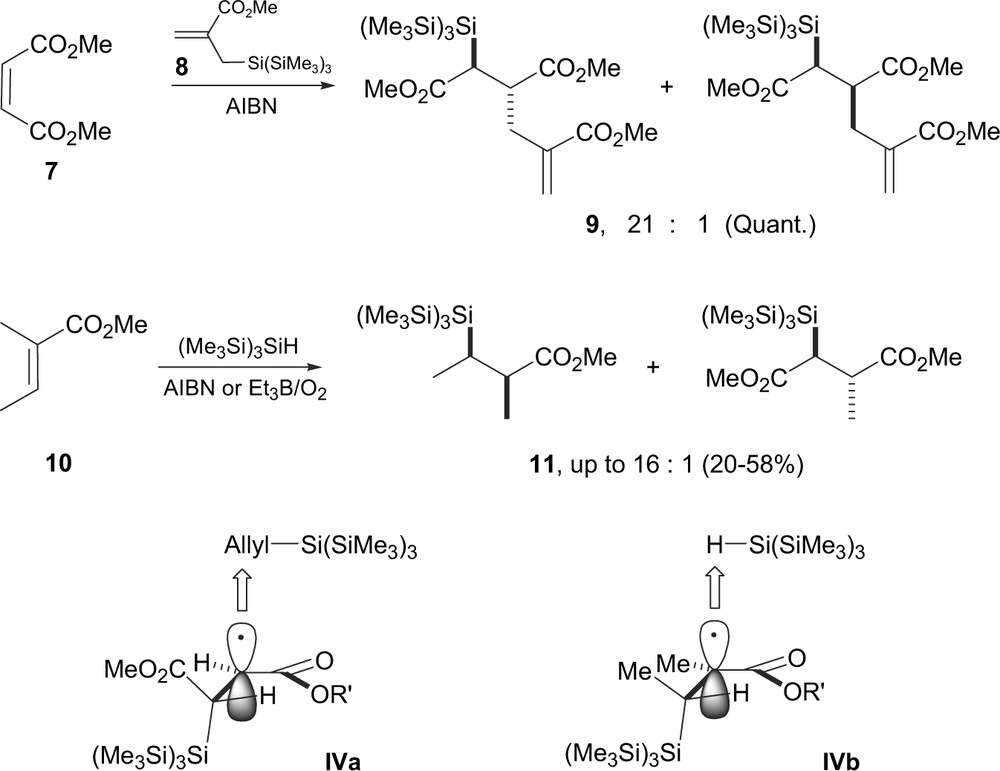
It is noteworthy that in the cyclic series, reduction of the anhydride 12 leads to the thermodynamically less stable cis isomer 13, formed as a unique diastereomer (Scheme 3) [13]. Similarly, allylation with 8 led to trans-14. Again, this follows prior observations by Bulliard et al. [9] who stated that as in maleic anhydride, both carbonyl groups are forced into a cis-arrangement, the bulky silicon group is then located on the opposite diastereotopic face of the radical as compared to the acyclic series. Approach of the allylsilane 8 or (Me3Si)3SiH would thus occur anti relative to the silicon group leading to 14 and 13, respectively (i.e. V, Scheme 3).
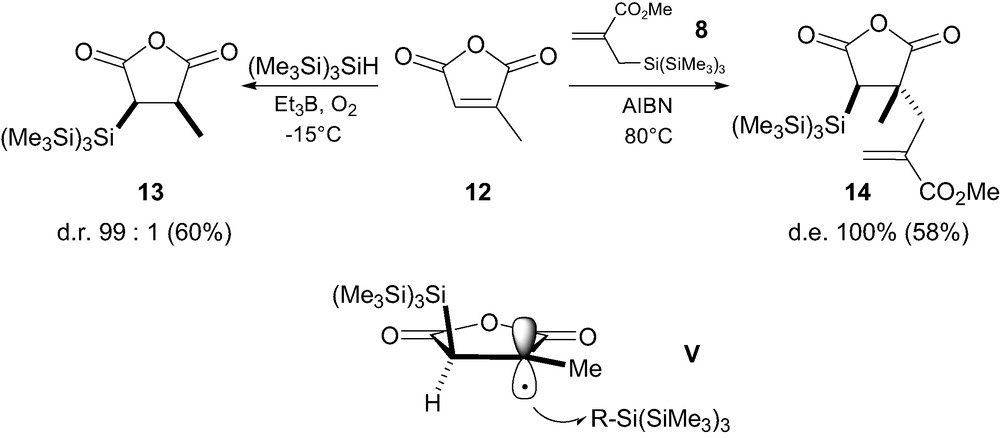
The presence of an ester function adjacent to the radical center unambiguously introduces a strong allylic strain favoring the conformation in which the smallest group in nearly in the same plane than the ester group, the large group being orthogonal to the π-bond, also minimizing A1,2 interactions (i.e. as in IVb) [7]. A different situation is visible in reactions involving acrylonitrile, where the linear substituent replaces CO2R. No conformation experiences allylic strain in this case so that attack from both faces of the radical leads to transition state of equal energy and no stereoselectivity is observed [13].
2.2 Felkin–Anh-type models
2.2.1 Deuteration of β-silylsulfinyl radicals
The allylic strain model above has been applied to esters but also to amides, ketones, amines and phenyl substituted radicals. The situation is different when no or weak stabilization of the radical occurs. In such cases, reversal of the sense of the diastereoselectivity is generally observed and the selectivity may be rationalized invoking a Felkin-Anh type model [14]. During our investigations on radical deuterations of α-sulfinyl-α-selenyl β-silanes 15 and 17 [15], we observed reasonable levels of stereocontrol, with the syn–syn diastereomer 16 formed majoritarely, from the syn-β-silyl sulfoxide 15, while the syn–anti stereoisomer 18a was formed starting from the anti precursors 17a (Scheme 4). These results were rationalized using a Felkin–Anh-type model VI, assuming that in their lowest energy conformation, these β-silyl radicals had a C–Si bond in the plane of the radical orbital, orthogonal to the C–S–O system and a S=O bond orthogonal to the SOMO [16], the sulfinyl group being weakly stabilizing. Calculations have shown that such radicals are quasi planar as represented in models VIa–b [17]. The main interactions would thus be those existing between the Ar group on the sulfinyl moiety and the bulky silicon group. In such conformations, interactions between SiMe2Ph and Ar and those between Bu3SnD and R would predominate, with the reagent Bu3SnD approaching anti relative to the bulky silicon group.
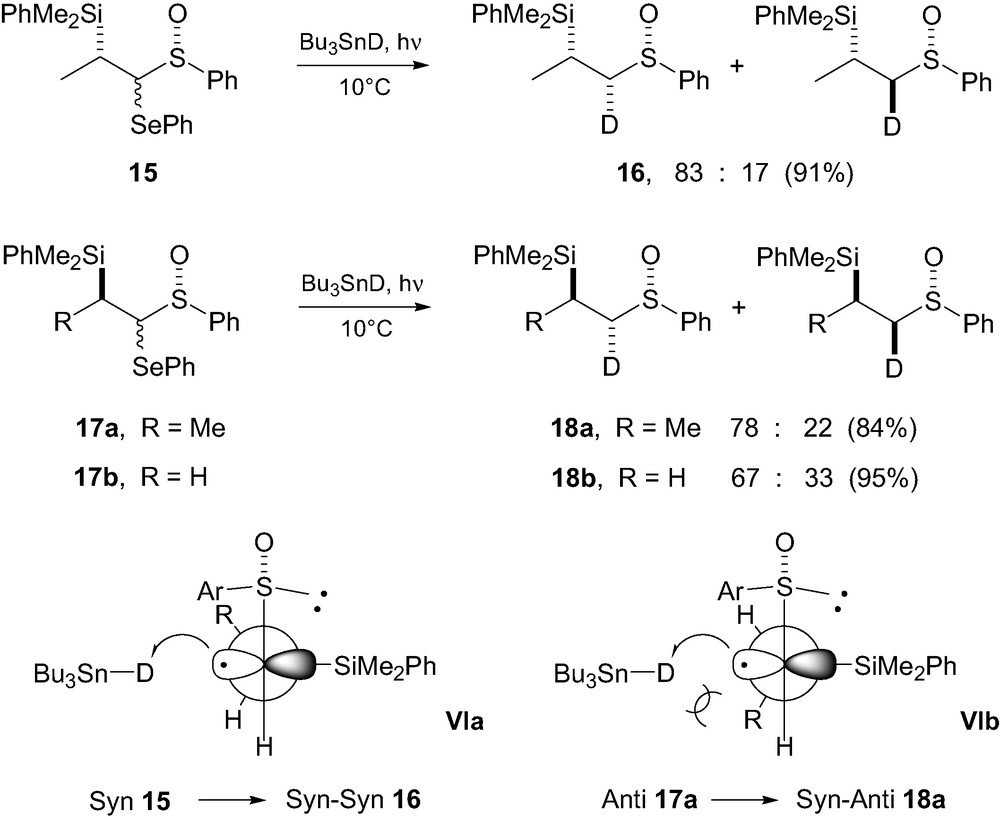
The slightly lower selectivity observed with anti 17a could be rationalized by the non-bonding interaction occurring between R and Bu3SnD in transition state VIb. Interestingly, radical deuteration of the thioether analogue 19a (Scheme 5) led to a much lower stereocontrol, indicating that in β-silylsulfinyl radicals, both stereogenic centers influence the stereochemical outcome and that the cooperativity between the chirality at sulfur and the chirality at C-β overwhelms the expected match and mismatched effects. This was further confirmed by the low selectivity observed upon deuteration of sulfoxide 17b, lacking the second stereogenic center bearing the silicon group. Deuteration experiments were also extended to the sulfone analogues 19b, which led to no stereocontrol. In the thioether and sulfone cases, the low and absence of stereoselectivities, respectively, have been rationalized using again a Felkin–Anh-type model VII (Scheme 5) with Bu3SnD approaching anti relative to the bulky silicon group on the side of the smallest substituent H. With the larger sulfonyl group, a gauche interaction with the Me group on the stereogenic center disfavors conformation VII, so that the approach of the deuteride may occur on the other side. Calculations have shown that stabilization of the radical center by a sulfur substituent decreases on oxidation to the sulfinyl and sulfonyl states, so that a delocalized radical species as with the esters analogues discussed above is unlikely [17].
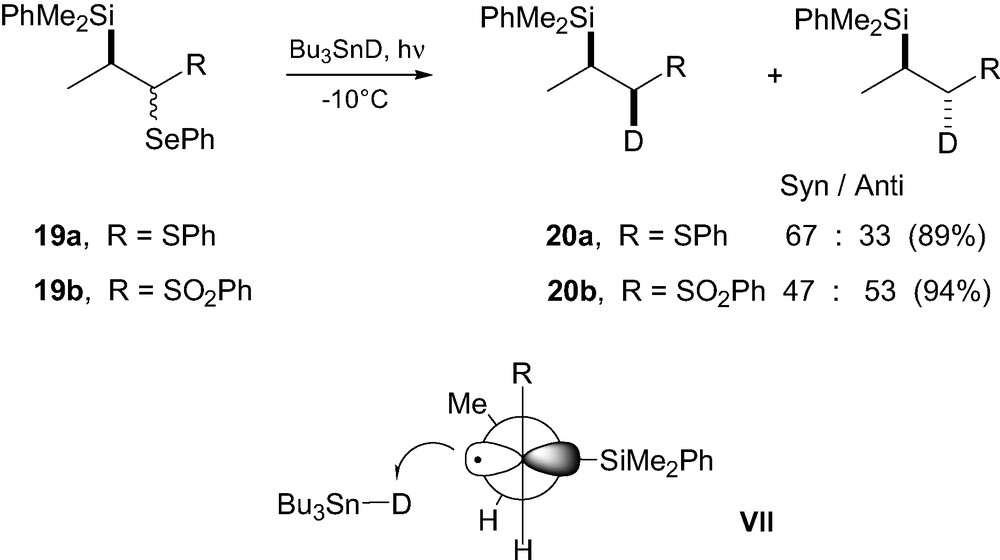
2.2.2 Allylsilanes in atom-transfer processes
Addition of free-radical species such as X to allylsilanes 21 generates, as an intermediate, a β-silyl radical species 22 which can react further with a radical trap (i.e. X–Y, Fig. 4) to lead to the addition product 23 [18]. Such a transformation has been reported for the first time by Topchiev et al. [19], and it was later demonstrated that allylsilanes reacted in atom-transfer processes with electrophilic radical species, five to seven times faster than non-silylated olefins [19b,20]. Such an enhancement of reactivity towards electrophilic radicals (i.e. CCl3) was at the time attributed to both (p–d)π homoconjugation and inductive effects [19b]. As the trichloromethyl radical is electrophilic in nature, the relative reactivity towards this species decreases with decreasing nucleophilic character of the olefin.
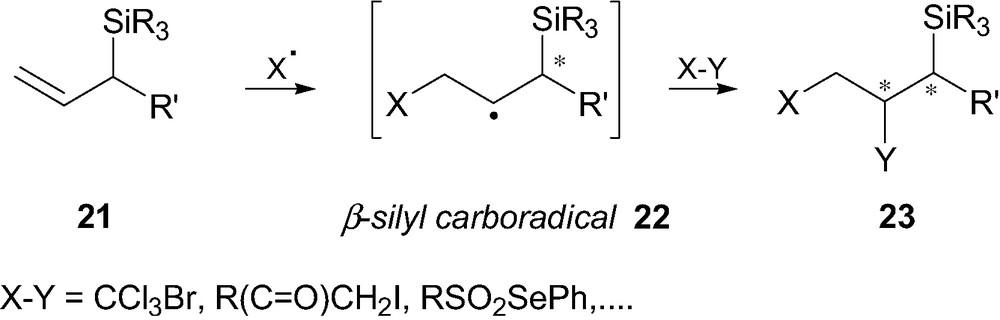
Allylsilanes in atom-transfer processes.
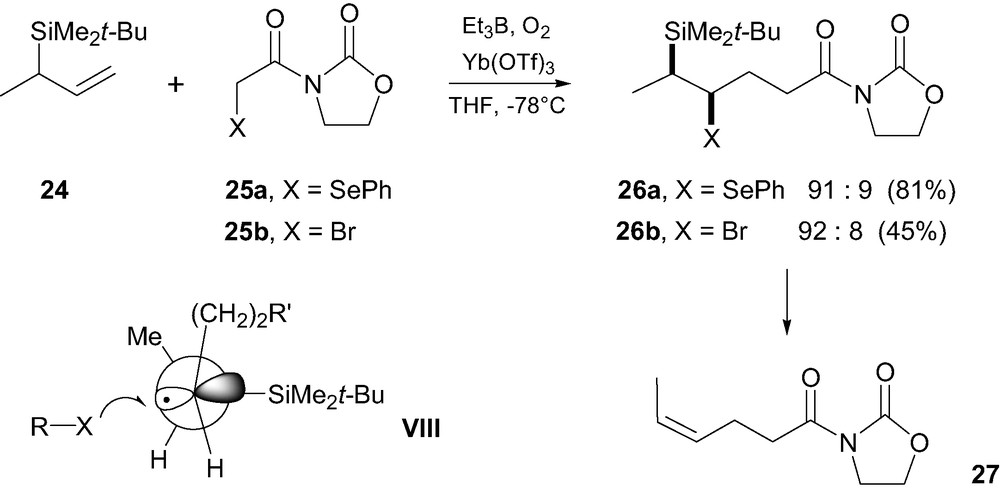
This strategy has recently found a renewed interest. Starting from a chiral allylsilane 24, Porter et al. [21] were able to show that Lewis-acid promoted addition, under free-radical conditions, of α-seleno and α-bromoimides 25a–b occurred with excellent levels of 1,2-stereocontrol, with a diastereomeric ratio of up to 94:6 in favor of the syn-isomer 26a–b (Scheme 6). A Felkin-Anh type model VIII was proposed to account for this 1,2-stereocontrol [21b]. Transfer of the bromo or the seleno group is believed to occur anti relative to the silicon group on the side of the smallest substituent H. β-Elimination of the β-seleno (or bromo)silane 26a–b led stereospecifically to the olefin 27 having a (Z)-configuration. Assuming an anti stereochemistry for this elimination, a syn-configuration was hence proposed for 26a–b. As far as we know, this is the first study on the 1,2-stereocontrol issued from radical functionalization of chiral allylsilanes.
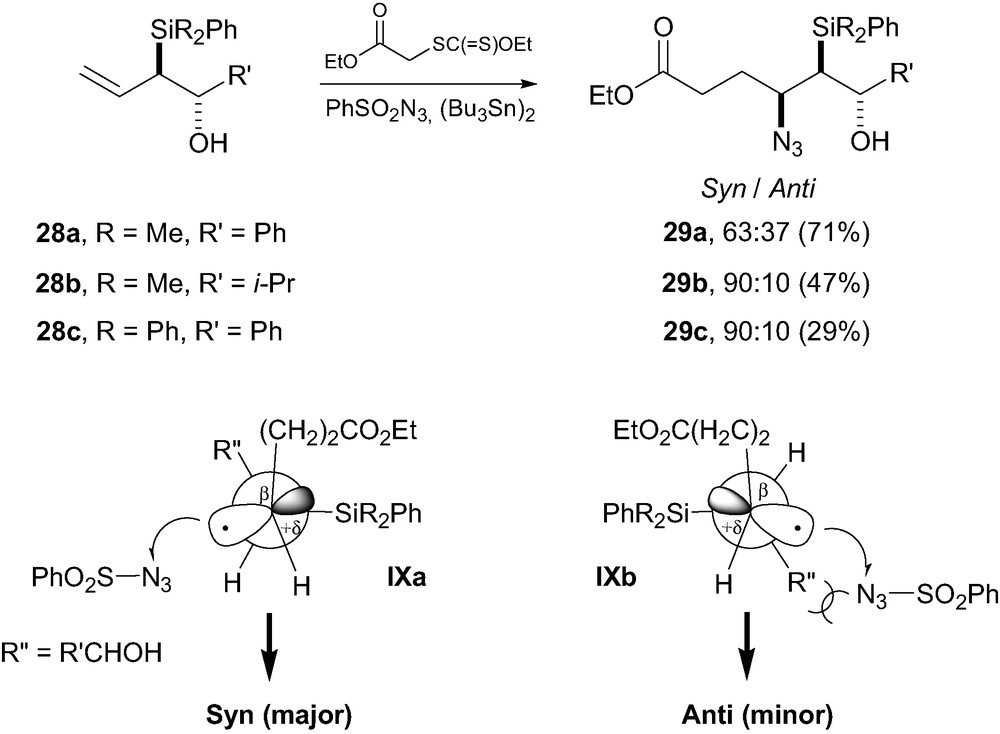
In line with these results, Chabaud et al. [22] described the first stereocontrolled carbo-azidation of chiral allylsilanes. Addition of xanthates onto a series of allylsilanes 28a–c possessing one or two stereogenic centers (in α and β-positions relative to silicon) led to the generation of a β-silyl carboradical intermediate that was trapped by a sulfonyl azide (Scheme 7). This led to the formation of β-azidosilanes 29a–c with a level of diasterecontrol ranging from 7:3 to 9:1. As mentioned above, fluoride-mediated elimination of the major isomers led exclusively to the (Z)-olefins. Based on a study on the stereochemistry of fluoride-mediated elimination of β-azidosilanes, [23] we were able to demonstrate that, similarly to Porter's atom-transfer reaction, the carbo-azidation led to the syn-isomers 29a–c as the major products. We rationalized our results by invoking a pyramidalization of the transition state into a quasi staggered conformation IXa to avoid gauche interactions between large groups (here between SiR3 and CH2CH2CO2Et groups) [24]. Such non-stabilized alkyl-substituted radicals are known to be pyramidalized in the ground state, and consequently the sulfonyl azide would approach anti relative to the bulky silicon group, on the side of the smallest group H. In the diastereomeric transition state conformation IXb, a larger steric interaction would exist between the sulfonyl azide and the medium-sized group R″. This is corroborated by the higher stereocontrol observed when R′ is i-Pr as compared with Ph. On the opposite when R″ is too hindered (i.e. R″ = i-PrCHOSiEt3), gauche interactions with the CH2CH2CO2Et group becomes important, leading to no diastereocontrol. Finally, as a consequence of the electrophilic nature of the sulfonyl azide, a partial positive charge is likely to develop β to the silicon group. This partial positive charge would be stabilized by the quasi-coplanar electron-rich C–Si bond (silicon β-effect), [2] thus adding to the intrinsic steric effect of the silicon group.
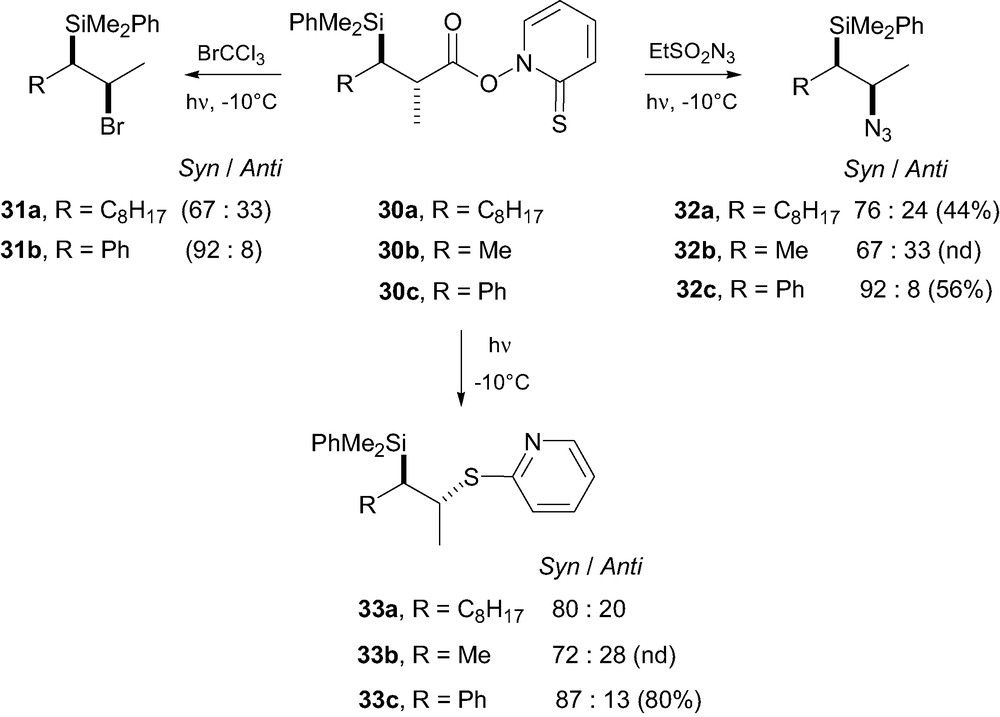
This transition state model is to be compared with A-strain TS model III (Scheme 1), [7] proposed for radical deuterations and allylations of closely related β-silylcarboradicals ‘conjugated’ with an ester. Clearly, allylic strain cannot play a role in this carbo-azidation process, and the Felkin-Anh type model IXa is in better agreement with our experimental results. A refinement of the models above and firm conclusions are expected to follow further experiments and high level calculations. Parallel to this work, Porter extended his studies on reactions of β-silyl carboradicals using different radical traps [25]. He was able to show that a general trend emerges from his and our studies i.e. that in all studied transformations, the syn-diastereomer is formed majoritarely. The β-silyl carboradicals were generated from Barton esters 30a–c of β-silylcarboxylic acids (Scheme 8). The latter were photolyzed in the presence of bromotrichloromethane or ethanesulfonyl azide leading to the corresponding β-bromo 31a–b and β-azido silanes 32a–c, respectively, in moderate to good yield with modest stereocontrol. The β-bromosilanes 31a–b are unstable and spontaneously undergo anti-elimination to produce the corresponding olefins. Rearrangement of the Barton esters 30a–c through photolysis also led to the corresponding thiopyridyl compounds 33a–c with the same level of stereocontrol as above. In each case, the syn-isomers were formed as the major compounds, which upon treatment with TBAF led to the corresponding Z-olefins. The best stereoselectivities were obtained when the medium-sized group on the stereogenic stereocenter was a phenyl. The stereochemical outcome of these different transformations can be rationalized as above, invoking the Felkin-Anh transition state model IXa, whatever the nature of the attacking species.
3 Stereocontrol in free-radical mediated 5-exo-trig cyclizations
As a continuation of our investigations on the free-radical functionalization of allylsilanes, we have recently studied the sulfonyl radical addition-5-exo-trig cyclization-β-fragmentation cascade onto allylsilanes 34a–b (Scheme 9) [26]. Addition of the electrophilic tosyl radical species onto the allylsilane moiety provides a β-silyl radical intermediate which then add in a 5-exo-trig fashion to the second olefin to provide the carbocycles 35a–b. Tri- and tetrasubstituted cyclopentanes 35a–b were thus obtained in excellent yields with unexpectedly high levels of diastereocontrol. Several related cyclizations were also conducted on analogues, bearing allylic methyl and hydroxy groups, to explore the generality of the method. It was eventually concluded that only a silicon group at the allylic stereogenic center was able to induce such high levels of stereocontrol. Reaction rates with allylsilanes were also much higher than those with non-silylated analogues, which were attributed to the electrophilic nature of pTolSO2 radical that reacts faster with electron-rich olefins such as allylsilanes.
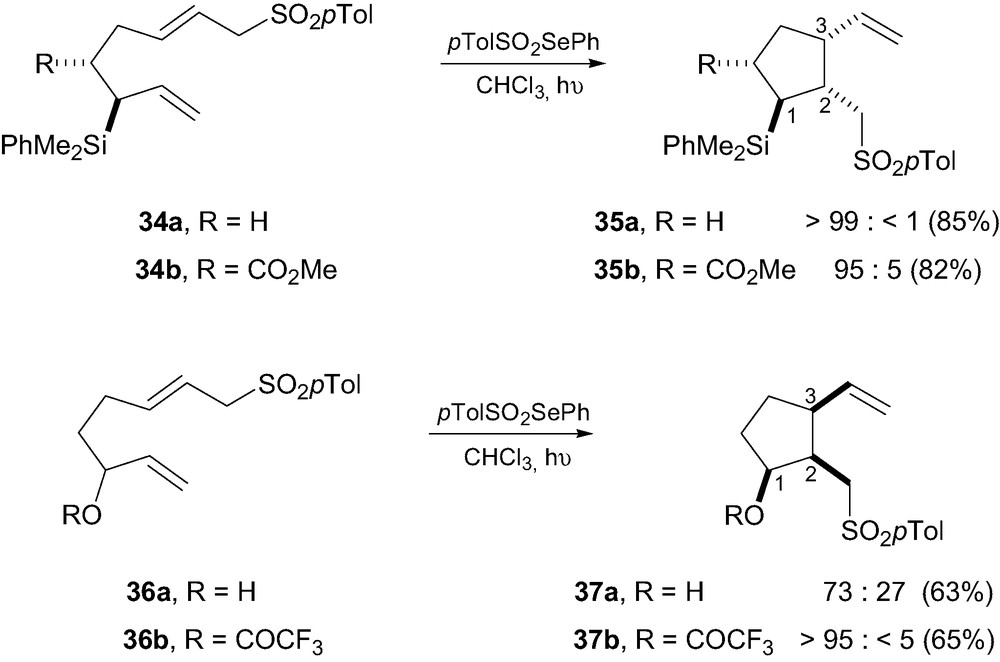
The stereoinduction was rationalized using the Beckwith-Houk model X (Fig. 5) [27]. The major diastereomer is likely to be formed through a chair-like transition state in which the bulky silicon group occupies a pseudoequatorial position. It is noteworthy that in such a conformation the electron-rich C–Si bond is nearly aligned with the incipient bond and can therefore stabilize the developing positive charge at C1, in β-position. This is reminiscent of the β-silicon effect discussed above for ionic processes [2]. Using this approach, it was possible to efficiently control the relative configuration between C1, C2 and C3 (1,2- and 1,5-stereocontrol), usually a difficult task to achieve. As far as we know, this is the first report on a successful control of the three contiguous stereogenic centers in such systems, using radical 5-exo-trig cyclizations. The same sequence applied to the alkoxy analogues 36a–b led to contrasting results [26]. Cyclization of diene 36a thus led to the cyclopentanes 37a having all substituents in a cis-arrangements. This was again rationalized using the Beckwith–Houk transition state models X, in which the OR group at C2 was located in pseudoaxial position in order to avoid a destabilization interaction with the developing positive charge at C1 [28]. The C1–H bond in pseudo-equatorial position would be better able to stabilize this native positive charge as it is a better σ-donor than a C–O bond. The better stereocontrol obtained with the model 36b having a strong electron-withdrawing CF3CO2 group at C2 further supports this hypothesis.
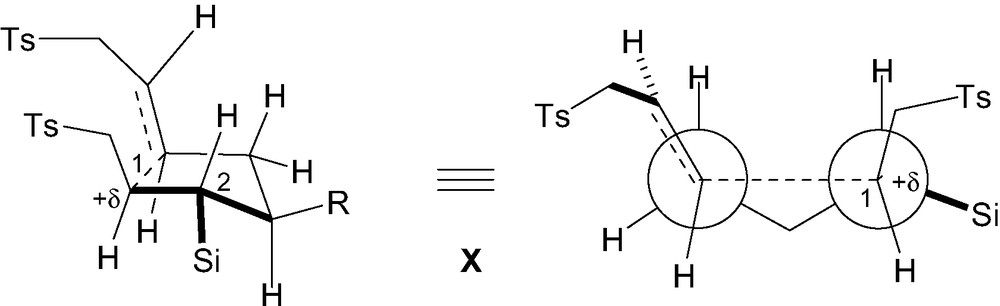
Stereochemistry of 5-exo-trig cyclization of dienes 34a–b and 36a–b.
4 Conclusion
We have described along these lines several recent examples of the transfer of chiral information occurring between a stereogenic center bearing a silicon group and an adjacent carboradical center. Generally, the stereoinduction is good and highly predictable on the basis of allylic and torsional strains. It is noteworthy that the models, developed earlier for ionic processes are also valid for these radical processes. The degree of pyramidalization of the radical species certainly introduced certain distortions in the existing models and should be taken into account.
Acknowledgements
I am grateful to the co-workers, whose names appear in the references, to the ‘Institut universitaire de France’ and the French ‘Ministère de l'Éducation et de la Recherche’ for financial support and finally to Professor P. Renaud (University of Berne, Switzerland) for fruitful discussions.