

1 Introduction
Group VI octahedral metal clusters of the form M6Q8L6 (M = Mo, W; Q = S, Se, Te; L = Lewis-basic organic ligand) have been the subject of much interest over the last few decades. Since these soluble, molecular clusters became accessible due to the work of Saito et al. [1,2] and McCarley et al. [3,4], research has been directed toward understanding their synthesis [5,6], electrochemistry [7,8], and ligand exchange chemistry [9,10]. One reason for the great interest in these cluster species has been their structural relationship to the fascinating molybdenum-based Chevrel phases, which are well-known for their superconductivity [11,12], fast-ion conductivity [13,14], and catalytic properties [15,16]. In addition, theoretical work [17,18] has shown that organic-inorganic network materials built from M6Q8L6 building blocks might also be expected to have interesting electronic and/or magnetic properties.
We have been interested in using ditopic nitrogen- and phosphorus-based ligands to build networks of covalently linked W6S8 clusters. This cluster core is inert except at high temperatures or under very basic conditions [19], while its axial ligands become labile under much more mild conditions, making ligand exchange a pathway to a range of W6S8-based compounds. Conceptually, construction of a network compound could be achieved by replacing the six monotopic ligands (L) on a molecular cluster with three equivalents of a ditopic ligand. Each bridging ligand would be shared between two clusters, leading to a three-dimensional network. In practice, this ditopic coordination has often produced insoluble, amorphous materials that are difficult to characterize [20,21]. We have also observed competing monotopic binding modes for ditopic ligands, which can result in molecular, zero-dimensional compounds. A Re6Se82+ cluster compound with monotopically bound 4,4′-bipyridine ligands has been prepared previously [22], but compounds with this mode of coordination have not been reported for the group VI clusters. The synthesis and characterization of two such compounds, W6S8L6 (L = bis(diphenylphosphino)ethane, 4,4′-bipyridine) is reported here, along with a discussion of the interplay between ditopic and monotopic binding modes for these two ligands.
2 Experimental
2.1 General
All manipulations were carried out inside an argon-filled dry box, unless otherwise indicated. Benzene, tetrahydrofuran (THF), and diethyl ether (Et2O) were distilled from sodium benzophenone before use. Acetonitrile (MeCN) was dried over 4 Å molecular sieves and distilled. Aniline was dried over potassium hydroxide pellets and distilled under dynamic vacuum. Deuterated solvents were purchased from Cambridge Isotopes Laboratory and used as received. W6S8(tbp)6 (tbp = 4-tert-butylpyridine) was prepared according to the literature procedure [5]. Bis(diphenylphosphino)ethane (dppe) was purchased from Strem, and 4,4′-bipyridine (4,4′-bipy) was purchased from Aldrich. Both ligands were used as received. The “reaction bomb” referred to below is a custom-made, thick-walled glass vessel equipped with a Teflon valve.
1H NMR spectra were collected on Varian Inova 400 and Bruker AF-300 spectrometers. Spectra were internally referenced against the residual protonated solvent peaks (δ 7.16 for benzene and δ 2.50 for DMSO). Powder X-ray diffraction was carried out on a Scintag XDS2000 diffractometer.
2.2 Synthesis of W6S8[bis(diphenylphosphino)ethane]6 (1)
A reaction bomb was charged with W6S8(tbp)6 (50 mg, 0.023 mmol), dppe (236 mg, 0.59 mmol, 25 equivalents), and benzene (8 g) to make a dark red solution. Outside the glove box, the bomb was heated in a 95 °C oil bath for 12 h, and the reaction mixture became a clear, orange–brown solution. The solution was divided among three vials and layered with approximately equal volumes of Et2O. Over a period of approximately 1 week, very small brown crystals formed at the sides and bottom of each vial. This solid was collected by filtration in air, washed with THF and Et2O, and dried in vacuo. The isolated product weighed 31 mg, a 30% yield. The crystalline powder was insoluble in all organic solvents tried, including benzene, DMF, DMSO, MeCN, aniline, and THF.
2.3 Synthesis of W6S8(4,4′-bipyridine)6 (2)
A reaction bomb was charged with W6S8(tbp)6 (0.200 g, 0.0922 mmol), 4,4′-bipy (2.77 g, 17.7 mmol, 190 equivalents), and aniline (15 g). The vessel was removed from the glove box and heated to 100 °C for 2 days, producing a dark red reaction mixture. The mixture was filtered through a syringe filter and then layered with MeCN (25 ml) to precipitate the product. The resulting reddish-black solid was collected by filtration and washed with Et2O. The isolated product weighed 0.153 g, a 60% yield. 1H NMR in DMSO-d6: δ 9.35 (d, 2, α-H), δ 8.77 (d, 2, β-H), δ 7.99 (d, 2, χ-H), δ 7.82 (d, 2, δ-H), all from bound 4,4′-bipy; δ 6.98 (m), δ 6.53 (m), δ 6.46 (m) and δ 4.99 (broad) from aniline; and δ 8.72 (d), δ 7.82 (d, overlapped with δ-H) from residual free 4,4′-bipy.
2.4 X-ray structure determination
Crystals of 1 suitable for single-crystal diffraction experiments were grown by layering the reaction solution described above with acetonitrile. Block-shaped, orange–brown crystals formed at the sides of the vial over a period of about 1 week. It was possible to grow block-like, dark red single crystals of 2 by layering acetonitrile on a diluted aniline reaction solution. Crystals were mounted on thin plastic loops using polybutene oil and immediately cooled in a nitrogen stream. Single-crystal diffraction data were collected on a Bruker SMART [23] system with a CCD detector using MoKα radiation. The cell parameters were initially determined using more than 50 well-centered reflections. The data were integrated using SAINT [23] software, and empirical absorption corrections were determined and applied using the SADABS [24] program. Space groups were determined through systematic absences and intensity statistics and the eventual ability to solve the structures. The structures were solved by direct methods using the SHELX [25] suite. Initial solutions, including positions of heavy atoms, were found using SHELXS, and positions of ligand atoms were determined using SHELXL to perform successive least-squares refinements on Fo2. Crystals obtained for 1 were small and somewhat weakly diffracting, and there was a particular lack of high-angle diffraction data. The final refinement employed a SHEL command line to limit the resolution to 1.0 Å, or 2θ = 41.6°. Anisotropic refinement was successfully applied to the tungsten, sulfur, and phosphorus atoms in 1 and to all non-hydrogen atoms in 2. Hydrogen atoms were added to the ligands using a riding model. Details of the crystal and refinement data for both molecules are shown in Table 1.
Crystal and refinement data for W6S8(dppe)6 (1) and W6S8(4,4′-bipy)6 (2)
| 1 | 2∙5aniline | |
| Empirical formula | W6S8C156H144P12 | W6S8C90H83N17 |
| Formula weight | 3749.93 | 2762.31 |
| Crystal system | Trigonal | Triclinic |
| Space group | R | P |
| a (Å) | 29.7159(13) | 9.3565(6) |
| b (Å) | 29.7159(13) | 15.7743(10) |
| c (Å) | 14.2188(9) | 15.8149(11) |
| α (°) | 90 | 99.5975(18) |
| β (°) | 90 | 98.4930(17) |
| γ (°) | 120 | 96.1659(18) |
| Volume (Å3) | 10873.5(10) | 2255.3(3) |
| Z, calculated density (g cm–3) | 3, 1.718 | 1, 2.034 |
| μ (cm–1) | 50.40 | 78.60 |
| F(000) | 5496 | 1314 |
| Crystal size (mm3) | 0.1 × 0.05 × 0.05 | 0.25 × 0.1 × 0.05 |
| Reflections collected/unique | 19,828/2529 | 22,372/9075 |
| Data/restraints/parameters | 2529/0/144 | 9075/0/550 |
| Goodness-of-fit on Fo2 | 1.159 | 1.033 |
| R1a (I > 2σ)/all data | 0.0474/0.0838 | 0.0367/0.0521 |
| wR2b (I > 2σ)/all data | 0.0920/0.1038 | 0.0712/0.0755 |
| Largest difference in peak and hole | 0.763 and –1.012 e Å–3 | 1.988 and –1.665 e Å–3 |
a R1 = Σ||Fo| – |Fc||/Σ|Fo|.
b wR2 = [Σw(Fo2 – Fc2)2/Σw(Fo2)2]1/2.
3 Results and discussion
3.1 Synthesis
The synthetic schemes used to prepare 1 and 2 are shown in Fig. 1. In both cases, a large excess of the ditopic ligand was used in order to assure the formation of molecular clusters and not cross-linked solids. The orange–brown reaction solution of 1 was clear, without any solid that would indicate formation of a linked product. The powder that was isolated by layering this reaction solution with Et2O was surprisingly insoluble, given the initial clarity of the reaction solution. Many equivalents of excess dppe were present in the reaction solution, which could lead to increased solubility for the mixture as compared with the pure product. However, adding excess ligand to the isolated product did not increase its solubility in benzene, nor did heating the suspension. This solid has, however, been found by X-ray characterization (vide infra) to be the molecular W6S8(dppe)6 product and not a linked product that might be expected to have low solubility. For the synthesis of 2, the reaction mixture did contain some insoluble particles that were separated by filtration prior to crystallization of the product. Using a very large excess of 4,4′-bipy in the reaction was found to minimize formation of these impurities. W6S8(4,4′-bipy)6 was alternatively synthesized from W6S8(tbp)6 in a slurry of 4,4′-bipy (75 equivalents per cluster) and toluene. The mixture was heated in a sealed tube to 125 °C and then slowly cooled to produce black needles that were determined crystallographically to be 2·2(4,4′-bipy).

Synthetic schemes for W6S8(dppe)6 (1) and W6S8(4,4′-bipy)6 (2).
For each reaction shown in Fig. 1, a range of stoichiometries was explored in order to characterize the interplay between monotopic and ditopic ligand binding modes. At high ditopic ligand:W6S8(tbp)6 ratios, 4,4′-bipy and dppe behave in the same way, leading exclusively to molecular, non-linked cluster species. At these stoichiometries, the competition between monotopic and ditopic binding modes is won by monotopic binding. The reaction to produce 1 was run with 25, 50, and 100 equivalents of dppe, resulting in clear reaction solutions with no evidence of precipitation. X-ray characterization of powders and/or single crystals of the reaction products showed that the monotopically coordinated product had formed. For ligand exchange involving 4,4′-bipy, formation of non-linked clusters was found to occur with the use of 20 or more equivalents of the ligand per cluster. The much larger excess (190 ligands per cluster) used in the reaction to produce 2 was found to be helpful in obtaining reproducible yields and avoiding formation of insoluble impurities.
When only a small excess of dppe or 4,4′-bipy was used in ligand exchange reactions with W6S8(tbp)6, the two ligands were found to behave in markedly different ways. When 3–6 equivalents of 4,4′-bipy are heated with W6S8(tbp)6 at 100 °C in benzene, the color changes from dark red to black within minutes of the start of heating as an amorphous compound falls out of solution. This compound cannot be recrystallized from any simple solvent system but has been determined to contain W6S8 clusters linked by ditopically bound 4,4′-bipy ligands [20]. By contrast, a 3:1 stoichiometric mixture of dppe and W6S8(tbp)6 was found to maintain its dark red color for at least 1 h after the start of heating. Even after 14–18 h of heating, the solution color had changed from red–brown to orange–brown, but everything remained in solution, without a hint of precipitation. The same was observed for a 1:1 stoichiometry. For dppe, the monotopic binding mode appears to win out over ditopic binding even in the presence of a low concentration of the ditopic ligand. In addition to differing from the behavior of 4,4′-bipy, this differs from the behavior of bis(diethylphosphino)ethane (depe), which also quickly produces an insoluble, amorphous solid when reacted with W6S8(tbp)6 in a 3:1 ratio [20]. Kinetics of substitution by the bulky dppe ligand might be expected to be slower than for the less bulky 4,4′-bipy and depe ligands. However, the almost immediate formation of amorphous products in reactions involving 4,4′-bipy and depe suggests that the linking reaction happens quickly, and some precipitate would certainly be expected after heating a reaction mixture including dppe for 18 h. The relative proximity of the two Lewis base centers on dppe is unlikely to be the origin of this behavior since one would expect similar behavior for depe, in which the two phosphine sites are also separated by an ethyl group. In fact, precipitates readily form in the case of depe. Another possible explanation is that when a dppe ligand is monotopically bound to the W6S8 cluster, the remaining available phosphine site is made inaccessible to the approach of other clusters by the interaction of the bulky phenyl rings on both phosphorus atoms and on neighboring ligands. This idea is somewhat supported by the crystal structure of the W6S8(dppe)6 molecule (vide infra).
3.2 Single-crystal structures
Both W6S8(dppe)6 (1) and W6S8(4,4′-bipy)6 (2) contain the W6S8 core, consisting of an octahedron of tungsten atoms face-capped by sulfur. ORTEP [26] representations of the asymmetric and molecular units of 1 are shown in Figs. 2 and 3, respectively. The asymmetric and molecular units of 2 may be seen in Figs. 4 and 5. Each dppe ligand in 1 is coordinated to a tungsten site through one of its phosphorus atoms. Ligands do not project straight outward from the cluster because of the 111° bends in the ethyl groups connecting the two phosphorus sites. Outer phosphorus atoms – those not coordinated to the cluster – are in close proximity to phenyl groups of other ligands on the same cluster. The crowding of these free phosphorus sites is more obvious in a space-filling model (Fig. 6). This is in contrast with the structure of 2, in which the rigid 4,4′-bipy ligands project straight out from the cluster in each direction. Although the situation could be somewhat different in solution, this may help to explain why dppe is not as apt to bind ditopically to two clusters as is 4,4′-bipy.
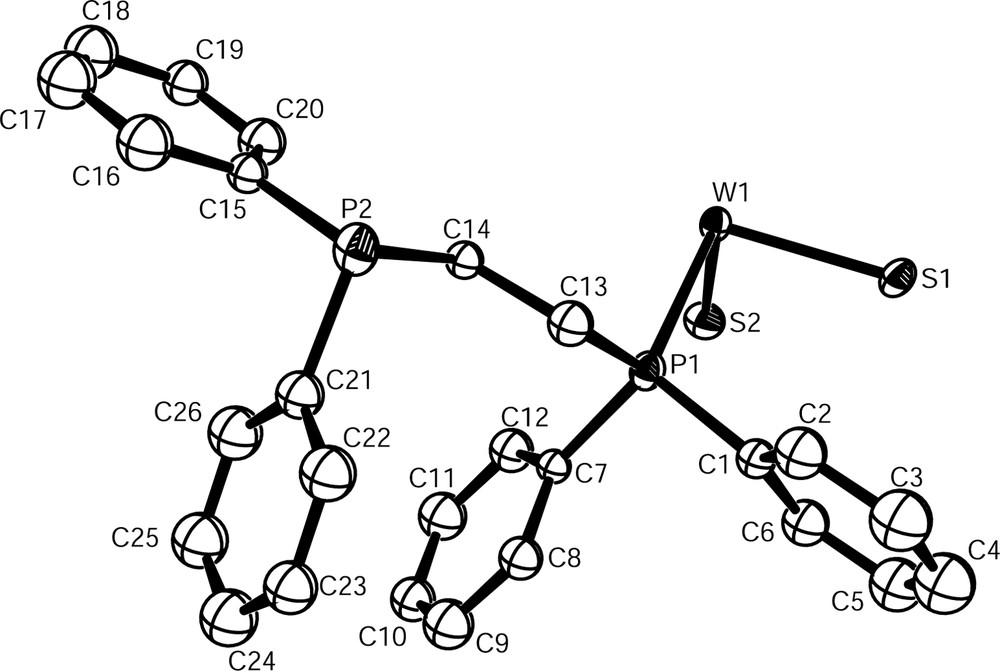
Asymmetric unit of W6S8(dppe)6 (1) with thermal ellipsoids drawn at 50% probability.
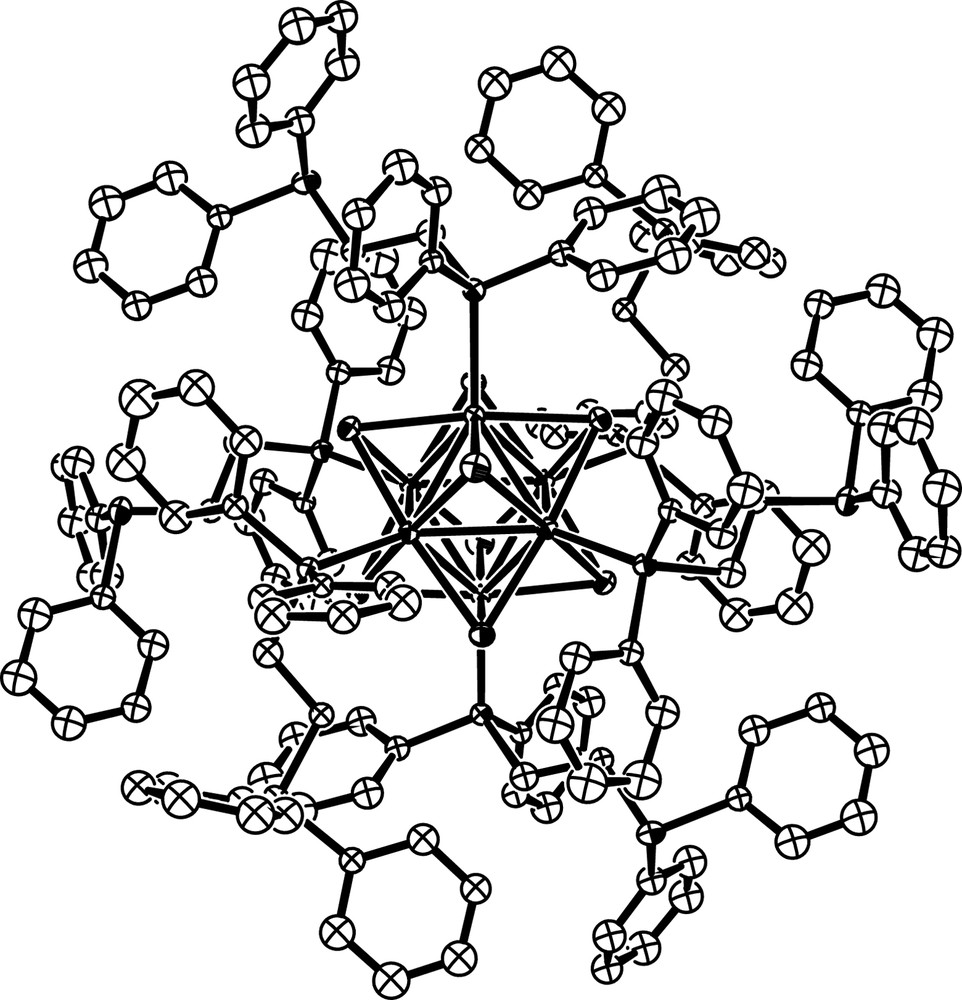
Molecular unit of W6S8(dppe)6 (1).
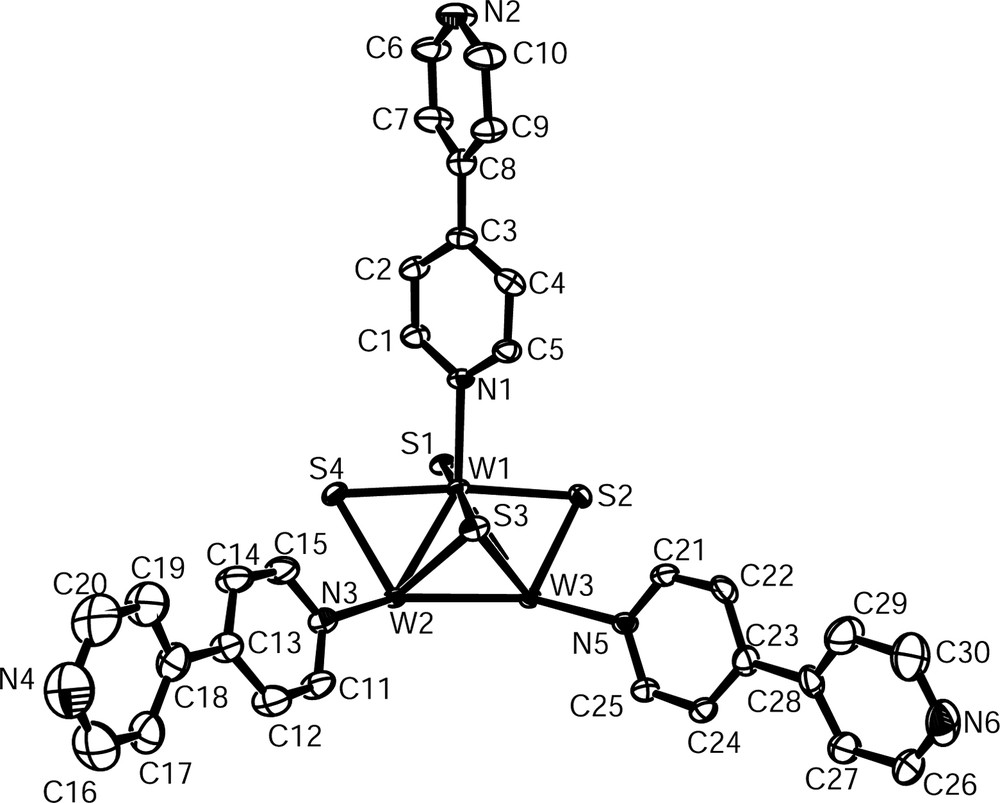
Asymmetric unit of W6S8(4,4′-bipy)6 (2) with thermal ellipsoids drawn at 50% probability.

Molecular unit of W6S8(4,4′-bipy)6 (2).

Space-filling model of W6S8(dppe)6 (1). Color scheme: C (gray), P (purple), W (black), S (yellow).
Select bond length averages and ranges for 1 and 2 are shown in Table 2 in comparison with the corresponding values for three previously synthesized W6S8 compounds. The tungsten octahedra in all of these compounds are quite regular, and W–S distances within the cluster core do not vary much among the compounds. Larger differences may be noted in W–W and W–L distances in comparing clusters with phosphorus- and nitrogen-based ligands. The W–P bond lengths are longer than the W–N bond lengths, and the W–W distances in the phosphine compounds are longer than those in the compounds with nitrogen-based ligands. This is consistent with what has been observed within an even larger group of W6S8 compounds [10]. For this group of compounds, bond orders were calculated to be greater for W–P bonds than for W–N bonds, and the larger W–W distances (and smaller bond orders) observed in phosphine compounds were attributed to the need to maintain a constant valence sum for tungsten.
Selected bond length ranges and averages (Å) for 1 and 2 in comparison with known W6S8 compounds
| 1 | 2∙5aniline | W6S8(PEt3)6a | W6S8(PPh3)6b | W6S8(tbp)6c | |
| W–W range | 2.6802(10)–2.6817(10) | 2.6581(3)–2.6685(4) | 2.6759(6)–2.6813(6) | 2.6749(4)–2.6899(4) | 2.656(1)–2.667(1) |
| W–W meand | 2.681(1) | 2.663(3) | 2.679(3) | 2.683(4) | 2.662(6) |
| W–S range | 2.435(5)–2.456(4) | 2.4372(17)–2.4637(15) | 2.451(2)–2.459(2) | 2.4262(18)–2.4692(19) | 2.458(2)–2.469(2) |
| W–S meand | 2.441(9) | 2.450(8) | 2.456(3) | 2.449(12) | 2.461(5) |
| W–L range | 2.240(5)–2.263(5) | 2.5561(19)–2.5915(19) | |||
| W–L meand | 2.543(4) | 2.251(9) | 2.523(2) | 2.570(12) | 2.257(8) |
Within the group of phosphine-ligated compounds, the bond lengths for 1 are similar to those reported for W6S8(PPh3)6 (PPh3 = triphenylphosphine) [10] and for W6S8(PEt3)6 [20]. In the past, the relatively large W–L distance of 2.570 Å for W6S8(PPh3)6 has been attributed to the bulkiness of the ligand that may prevent closer association with the cluster [10]. By contrast, W6S8(PEt3)6 has a smaller W–P distance of 2.523 Å. The phosphine binding site of dppe is more sterically hindered than that of PEt3, but the presence of two phenyl groups instead of three makes it more accessible than that of PPh3. Thus, it is not surprising that the W–L distance for 1, 2.543 Å, lies midway between the values for the other two compounds. The W–W and W–L distances in 2 are within experimental uncertainties of the distances previously found in W6S8(tbp)6 [27].
A view of the unit cell packing for 1 as viewed along the c-axis is shown in Fig. 7. Clusters are centered on the cell vertices and on the (1/3, 2/3, 2/3) and (2/3, 1/3, 1/3) special positions. Because of the roughly spherical shape of the molecular unit of 1, it is not surprising that this compound crystallizes in the R space group. This space group has also been found for other W6S8 cluster compounds, including W6S8(tbp)6 [27] and W6S8(PEt3)6 [20]. A structure was also found for a single-crystal of 1 grown by layering of the reaction solution with Et2O. The cell parameters for this structure differed slightly from the parameters reported in Table 1, with a = b = 29.8064 Å and c = 14.2891 Å. Arrangement of clusters within the unit cell was the same as for the structure reported here. However, this crystal was more disordered than a crystal grown by layering with acetonitrile, and the final R-values were higher.
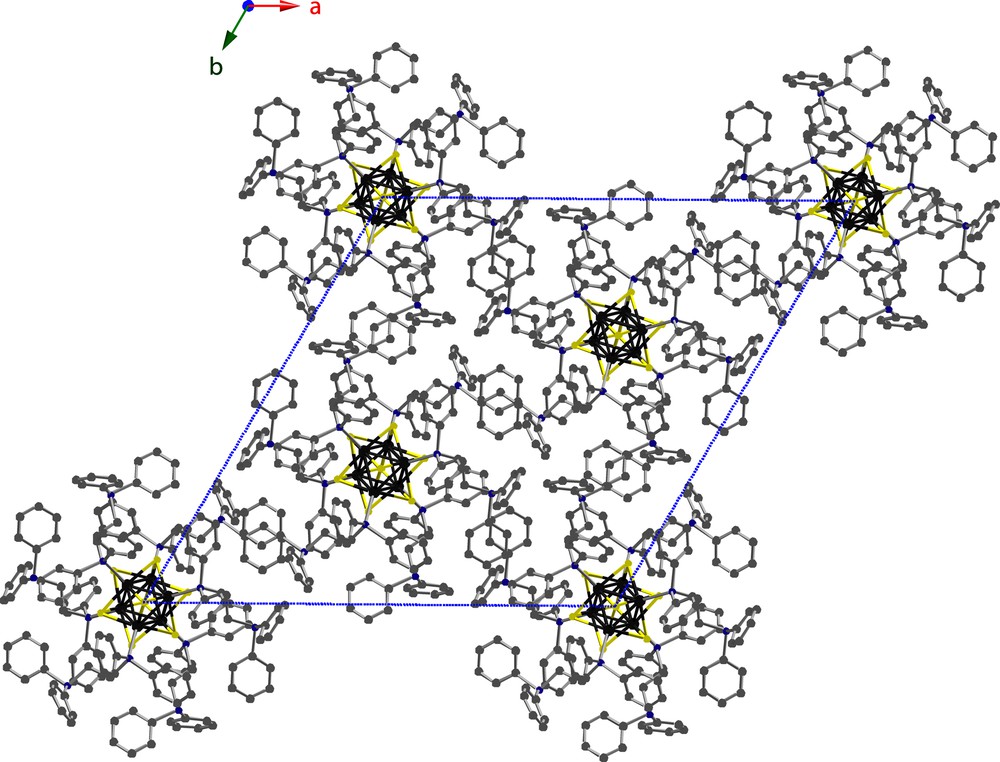
Unit cell packing of W6S8(dppe)6 (1), viewed along the c-direction.
The unit cell packing for 2 is shown in Fig. 8. Clusters are situated on –1 special positions. Aniline molecules of crystallization occupy the channels among clusters. A structure was also found for the 2∙2(4,4′-bipy) compound grown from a 4,4′-bipy/toluene melt. These needle-like crystals were also found to be in the P-1 space group, with a = 9.4544(11) Å, b = 14.6207(17) Å, c = 15.8388(18) Å, α = 67.703(3)°, β = 81.334(3)°, and γ = 86.142(3)°. The cell axes of this structure and that of 2∙5aniline agree within 10%, although there are bigger differences in the cell angles. Clusters are centered on the same special positions in both structures, with the main difference being the identity of the molecules of crystallization.

Unit cell packing of W6S8(4,4′-bipy)6 (2), viewed approximately along the a-direction.
3.3 Powder X-ray diffraction
The insoluble W6S8(dppe)6 powder was characterized by powder X-ray diffraction. The diffraction pattern that was collected is shown in Fig. 9, along with a pattern simulated by PowderCell [28] using the structural information found by single-crystal diffraction. Since the bulk powder was obtained from a mixture of benzene and Et2O, the structural data used in simulating the pattern were from a single-crystal grown from this same solvent mixture. As mentioned above, these parameters are slightly different than those for a crystal grown from benzene/acetonitrile.
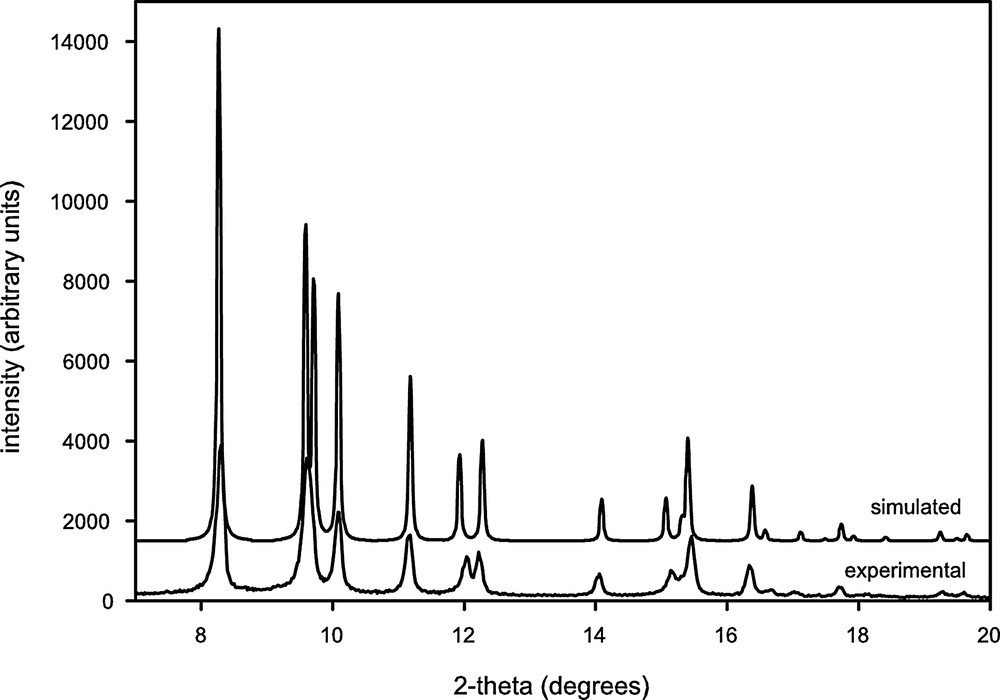
Overlay of simulated and experimental powder X-ray diffraction patterns for W6S8(dppe)6 (1).
JADE software [29] was used to find the peak positions in the experimental pattern, allowing a quantitative comparison between the experimental and simulated data. This comparison may be seen in Table 3. There is a systematic difference between the two sets of peak positions, with the simulated 2θ values an average of 0.082° larger than the experimental values. In addition to a possible contribution from zero-point error in the X-ray diffraction measurement, this difference may arise from the fact that the powder pattern was collected at room temperature, whereas the single-crystal data were collected at 173 K. The sign of the difference that is observed is consistent with what would be expected for a small expansion of a unit cell upon warming from 173 K to room temperature.
Calculated and observed powder X-ray diffraction peak positions (degrees 2θ) for W6S8(dppe)6 (1)
| h k l | Experimental | Simulated | Difference (sim – exp) |
| 1 1 0 | 5.905 | 5.926 | 0.021 |
| 1 0 1 | 6.986 | 7.065 | 0.079 |
| 2 0 –1 | 9.143 | 9.226 | 0.083 |
| 3 0 0 | 10.249 | 10.272 | 0.023 |
| 1 2 –1, 2 1 1 | 10.905 | 10.972 | 0.067 |
| 2 2 0 | 11.833 | 11.867 | 0.034 |
| 1 0 –2 | 12.712 | 12.846 | 0.134 |
| 3 1 –1, 1 3 1 | 13.750 | 13.824 | 0.074 |
| 4 0 1 | 14.980 | 15.052 | 0.072 |
| 2 1 –2, 1 2 2 | 15.220 | 15.359 | 0.139 |
| 4 1 0, 1 4 0 | 15.655 | 15.720 | 0.065 |
| 3 2 1, 2 3 –1 | 16.114 | 16.189 | 0.075 |
| 1 3 –2, 3 1 2 | 17.386 | 17.523 | 0.137 |
| 4 0 –2 | 18.367 | 18.512 | 0.145 |
| Average difference = | 0.082 |
The program Latcon-X [30] was used to determine unit cell parameters using the experimentally observed values of 2θ and the hkl indices obtained from the simulated pattern. This refinement found a = b = 29.9110 Å and c = 14.4608 Å. As compared with the values for a single-crystal grown from benzene/diethyl ether, these parameters show increases of 0.104 and 0.172 Å for a and c, respectively. Returning to Table 3, it may be observed that the largest differences in peak positions correspond to reflections for which l is large. This is consistent with the finding that Δc/c differs more than Δa/a in a comparison of cell parameters from powder and single-crystal data.
Qualitatively, intensities are in fairly good agreement between the two patterns. Some differences may result from preferred orientation of crystallites in the powder sample. The good agreement between simulated and experimental patterns helps to confirm the identity of the bulk powder in the absence of NMR data.
4 Conclusions
Two new compounds, W6S8(dppe)6 (1) and W6S8(4,4′-bipy)6 (2) were synthesized through ligand substitution on the W6S8 cluster core. Both of these ligands are ditopic and could potentially coordinate to two different clusters, but the structures found here show these ligands in monotopic binding modes. Use of 4,4′-bipy leads to this monotopically bound product when a large excess of the ligand is used but produces an amorphous, insoluble product when a small ligand: cluster stoichiometry is employed, presumably due to linking of clusters. Dppe behaves rather differently than 4,4′-bipy in that it does not appear to link clusters even when a low stoichiometry of the ligand per cluster is used. This suggests that the second phosphorus site is inaccessible to a second cluster due to the bulkiness of the phenyl groups and of surrounding ligands. Both compounds have been characterized structurally through single-crystal X-ray diffraction, and a powder X-ray diffraction pattern collected for bulk material of 1 agrees well with a pattern simulated from a single-crystal structure.
The supplementary material has been sent to the Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB2 1EZ, UK (CCDC numbers 247494 and 247495) and may be obtained by contacting the CCDC.
Acknowledgements
We acknowledge Dr. Craig Downie and Dr. Emil Lobkovsky for assistance with single-crystal diffraction experiments and the US Department of Energy (DE-FG02-87ER45298) for financial support. C.M.O. thanks the US National Science Foundation for Graduate Research and Graduate Teaching Fellowships (DUE# 0231913 and 9979516).