

1 Introduction
13C NMR spectroscopy offers a high potential for the quantitative analysis of complex mixtures, due to the broad spectral range and, usually, an absence of interference between peaks. It suffers, however, from poor sensitivity due to the low abundance and small magnetic moment of the 13C nucleus. This sensitivity is further diminished if the conditions of quantitative NMR − inverse-gated-decoupling procedure [1] and a recycling delay between 5- and 10- times the longest spin–lattice relaxation time [2–4] – are employed. In fact, the use of these acquisition conditions may lead to unacceptably long accumulation times to obtain a spectrum with an acceptable signal/noise ratio for a solute in small concentration. The alternative is to add a paramagnetic relaxation reagent [5], such as triacetylacetonatochromium (III) Cr(AcAc)3, which shortens the relaxation times. The unwanted side-effect of adding Cr(AcAc)3 is that it leads to line-broadening. Furthermore, adding a paramagnetic relaxation reagent may alter the compound under investigation. Alternative methods have been employed, detecting the signal in non-quantitative conditions for comparing concentrations of molecules with similar relaxation properties [6]. Also, the DEPT sequence [7] can be applied for quantitative analysis when optimised [8], but only to compare carbons of the same nature, which resonate in a small spectral range. Finally, when an accurate 13C NMR quantitative spectrum is needed for a sample with different carbons in a 200 ppm range, the inverse-gated-decoupling sequence is still employed with a recycling delay of about seven times the larger T1, for the different carbons of interest [9]. We propose here to improve the inverse-gated-decoupling sequence so that the recycling delay may be significantly reduced while the results remain quantitative.
2 Theory
2.1 General description of the magnetisation time course during the recovery delay
We consider a simple IS spin system (I = insensitive: 13C; S = sensitive: 1H), coupled by intramolecular dipole–dipole relaxation. All the development will be done for 13C and 1H, but similar results could be deduced for other nuclei.
The standard inverse-gated-decoupling sequence for quantitative measurements has been developed by Freeman et al. [1]. It is [T − Don – 90° − FID – Doff]n, where Don and Doff symbolise the switching of the proton decoupler, T is a waiting time allowing the system to return to thermal equilibrium, usually chosen between 5 and 10 T1(13C) [2–4].
During acquisition, I magnetisation is detected in the transverse plane while S is saturated for obtaining a decoupled spectrum. In the same time, 〈Iz〉 increases and would reach the steady state value I0(1+ η) if the acquisition time was very long. This may increase the delay T necessary for 〈Iz〉 to go back to I0 before the next detection. That is why the acquisition time TAQ is usually chosen short and at the end of the acquisition 〈Iz〉 = is shorter than I0, depending on the length of the acquisition time.
At the end of the acquisition, S is no longer saturated. The behaviour of I and S is described by the two differential equations, first derived by Solomon [10]:
| (1) |
| (2) |
| (3) |
| (4) |
Eq. (3) means that Iz goes back to equilibrium biexponentially. The relaxation rates λ+ and λ− are both functions of RI, RS and σ, with λ+ larger than λ−. This means that the corresponding exponential term exp[−λ+ t] should vanish more rapidly than exp[−λ− t].
Then, Iz returns to equilibrium more rapidly if C− << C+. In that case, equilibrium should be reached for t = 5/λ+.
It is interesting to determine the value of 〈Iz(0)〉 for which C− would become null. It is called :
| (5) |
This equation can be simplified in two extreme situations.
If σ >> |RS − RI|, λ+ − RI ≅ σ and ≅ I0 − S0.
If σ << |RS − RI|, λ+ ≅ RS. The nuclear Overhauser enhancement factor η is defined by the relationship:
| (6) |
In that case, can be written:
| (7) |
| (8) |
This shows that can vary in a wide range.
In most cases, it should be possible to adapt the NMR sequence so that at the beginning of the delay T, 〈Iz〉 = instead of . This would enable 〈Iz〉 to go back to equilibrium faster. The modification will depend on the value of compared to .
2.2 Optimisation of the sequence
Three cases can be considered.
(i) If < < I0, it is possible to saturate S during a delay Tsat after the acquisition until 〈Iz〉 = . In that case, Tsat is adjusted so that:
| (9) |
(ii) If − < < , • Iz• can be reduced to the desired value by applying a RF pulse of angle θ.
(iii)If − I0(1 + η) < < −, 〈Iz〉 could be amplified by saturating S after the acquisition until 〈Iz〉 = − . An inversion pulse would then enable one to obtain the desired value for 〈Iz〉.
Carbons with different relaxation parameters may be present in a same sample. In that case, a global optimisation could still be obtained by saturating S during a delay Tsat sufficient for increasing 13C magnetisation with the larger value of , then replacing the RF pulse of angle θ by series of selective pulses with adequate flip angles for each carbon.
The corresponding sequence is represented in Fig. 1 for two types of carbons. As this sequence consists in Inverse-Gated Optimised Decoupling, we propose to call it ‘InGOD’.
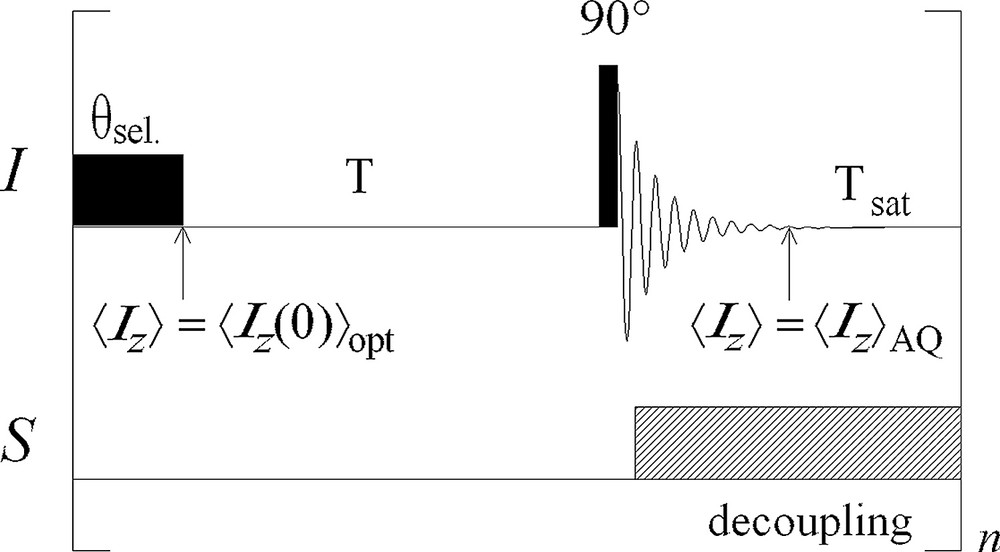
Inverse-gated-decoupling sequence optimised for a faster quantitative analysis. A supplementary delay Tsat and a selective pulse θsel are added at the end of the acquisition. The values of Tsat and θsel are adjusted so that 〈Iz〉 = at the beginning of the recovery delay T.
3 Material and methods
N,N-Dimethylacetamide, obtained from Sigma Aldrich, was diluted to 10% (v/v) in deuterated water and analysed in a 5-mm sample tube. All NMR spectra were recorded at 303 K, on a Bruker Avance 400 DPX spectrometer operating at 9.4 Teslas. 13C spectra were recorded at a spectrometer frequency of 100.62 MHz with a 5-mm dual probe. Free induction decays (FIDs) were accumulated in 64-K channels, with a spectral width of 19.38 kHz, an acquisition time of 1.69 s, using 90° read pulses of length 6.8 μs, and eight scans. Proton decoupling was obtained with the WALTZ16 mode. Selective pulses were obtained by applying an attenuated RF field during 133 μs. An exponential apodisation function of 5.0 Hz was applied to the FID prior to Fourier transformation. Phase and baseline were corrected manually. The areas of the peaks were measured by Lorentzian deconvolution with the Bruker program X-WinNMR 2.6. Nuclear Overhauser enhancement factors (η) for the different carbon sites were obtained by comparison of the corresponding peak areas Isat and I0 when 1H were decoupled and inverse-gated decoupled respectively [1], waiting 320 s between the end of the acquisition and the read pulse. They are defined as: Isat/I0 = 1 + η. The recovery of 13C magnetisation was studied by applying either an inverse-gated-decoupling sequence or the optimised sequence (Fig. 1) and waiting delays from 3 μs to 320 s before the read pulse.
Longitudinal relaxation times of 13C were determined by using an inversion–recovery sequence with the decoupler on continuously. An inversion–recovery sequence was employed for 1H longitudinal relaxation times determination. There, the baseline was corrected automatically and the areas of the peaks were analysed within the Bruker software.
4 Results and discussion
At the temperature considered, N,N-dimethylacetamide is composed of three non-equivalent CH3 groups and of a carbonyl group. As the relaxation properties of the two N–CH3 groups are quite close, we considered only three different spin systems: CH3–C, CH3–N and C=O. Their relaxation times are given in Table 1, together with their nuclear Overhauser enhancement η at steady state while 1H were saturated. For each carbon, the longitudinal relaxation time of the closer 1H group was also reported. The cross-relaxation coefficients σ were deduced from these values via Eq. (6). This enabled the calculation of λ+ via Eq. (3). Finally, the value of was calculated via Eq. (7) and is reported in Table 1. The value of 〈Iz〉 at the end of the acquisition was measured by applying an inverse-gated-decoupling sequence where the delay T is null. It is called and is also reported. From these results, and in order to increase the carbon relaxation, we have chosen to enhance all 13C magnetisation during a delay Tsat so that for C=O at the end of the delay Tsat. The length of Tsat was deducted from Eq. (9). It is 17.7 s. Then, the 13C magnetisation of the CH3 was significantly reduced by applying a selective 90° pulse at their average frequency. Fig. 2 represents the 13C magnetisation time course as a function of T for the optimised pulse sequence (■) compared to the standard inverse-gated-decoupling sequence (○) for the carbonyl group (Fig. 2a) and for a CH3 carbon bound to the carbonyl (Fig. 2b). Similar curves as in Fig. 2b were obtained for the other carbons CH3. One observes that, in both cases, magnetisation recovers faster during the delay T when the InGOD sequence is employed. For a given carbon, if one compares this delay T to the T1 of the corresponding carbon, it is in a range from 0.17 T1, for the carbonyl, to 3.7 T1, for the carbon C-2, which is much lower than the delay usually recommended for the inverse-gated-decoupling sequence. As a matter of fact, the additional delay Tsat makes the experiment longer. In order to assess the efficiency of our optimised version against the standard inverse-gated-decoupling sequence, we compared the global delay Tsat + T necessary for magnetisation relaxation to the larger relaxation time T1max (Table 2). Finally, the delay needed for complete recovery of the magnetisation of all carbons is 50.7 s, which is less than 1.8 T1max, thus much less than the delay of 7 T1max, usually recommended for allowing magnetisation of quaternary carbons to return to equilibrium after the inverse-gated-decoupling sequence [9].
Relaxation properties for the different carbons and for the protons dipolar coupled to them. Comparison of the magnetisation at the end of the acquisition to the optimal value deduced from the relaxation parameters
| Carbon | T1(13C) | T1(1H) | η | ||
| C-3 (C=O) | 28.8 s | 2.81 s | 0.85 | 0.18 | 0.91 |
| C-2 (CH3–N) | 9.04 s | 2.99 s | 1.63 | 0.59 | 0.22 |
| C-1 (CH3–C) | 8.62 s | 2.81 s | 1.70 | 0.63 | 0.21 |
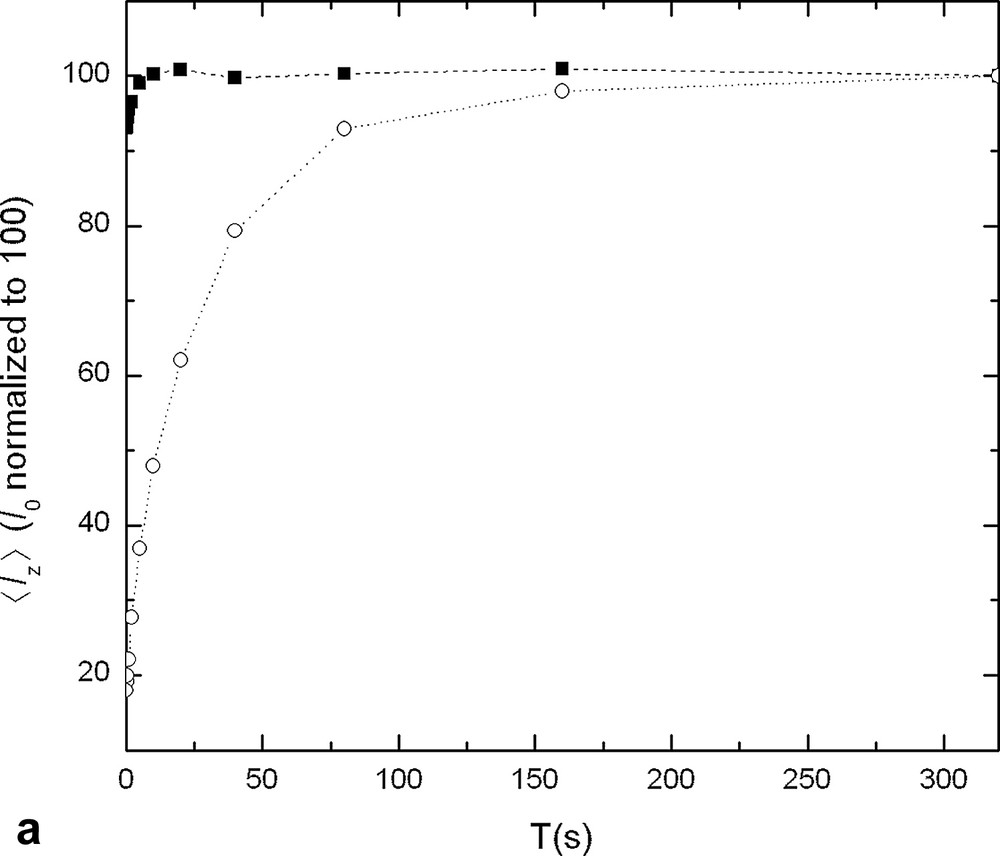
13C longitudinal-magnetisation time course as a function of the recycle delay T, for the carbonyl group (a) and for a CH3 carbon bound to the carbonyl (b). In the two graphs, the signals detected after the optimised sequence (■) are compared to those detected after the standard inverse-gated-decoupling sequence (○).
Recovery delay of N,N-dimethylacetamide 13C magnetisation after the saturating delay and the selective pulse for which 〈Iz〉 = I0 with a 1% precision
| Carbon | T | Tsat + T |
| C-3 (C=O) | 5 s | 22.7 s (0.79 T1max) |
| C-2 (CH3–N) | 33 s | 50.7 s (1.8 T1max) |
| C-1 (CH3–C) | 21.5 s | 39.2 s (1.4 T1max) |
One should note that for the sample studied, the quaternary carbon is well separated from the CH3 groups, which have similar relaxation properties and are in a small range. Therefore, it is very easy to apply a selective pulse on the CH3 groups without disturbing the carbonyl magnetisation. If protonated carbons would be in a more wide range, it should be possible to apply a series of selective pulses with optimal angles at the desired frequencies. For a better selectivity, shaped pulses as gaussians should then be used instead of low power rectangular pulses.
5 Conclusion
We have enhanced the inverse-gated-decoupling sequence for quantitative analysis to be performed faster. This optimisation can be performed accurately if the longitudinal relaxation times of the cross-relaxing nuclei are known, together with the NOE coefficients. It is particularly efficient in a sample containing quaternary carbons. The quantitative analysis of other carbons can be achieved provided that a selective pulse is applied at their resonance frequency. If this cannot be done, quantitative analysis could be performed in two separate experiments optimised for quaternary and fast relaxing carbons respectively. This method may be applied each time precise quantitative NMR is needed despite the presence of dipolar couplings.