

1 Introduction
The probing of the adhesion, tribological properties, and viscoelastic properties of polymer films have received great attention in recent years due to the importance in practical applications such as adhesives, sensors, microelectronics, coating, lubricants and liquid crystalline display and biomaterials [1]. Tribological properties of polymeric solids are closely related to the molecular motions and aggregation state at the surface. Due to the breaking of symmetry at the air–polymer interfaces, the polymer chain conformations and molecular thermodynamic properties could be quite different from those of the bulk polymers. For example, since the chain end group would preferentially segregate at the polymer/air interface as indicated by neutron reflectivity measurements [2], there should be more free volume associated with chain ends, and the glass transition temperature of the surface region should be less than that of the bulk which was also observed by Keddie et al. [3] using the ellipsometric measurements and by Kajiyama et al. [4] using temperature-dependent X-ray photoelectron spectroscopy (TDXPS) and angular-dependent XPS (ADXPS). In spite of some efforts, the detailed and consistent conclusions could not be reached so far. For example, using near edge X-ray absorption fine structure (NEXAFS), Liu et al. [5] found no evidence of enhanced mobility and the depression of the glass transition. Xie et al. [6] also drew the similar conclusion based on the positronium annihilation measurements.
In this paper, an ultra-thin film is defined as the film having thickness comparable to the radius of gyration, Rg, of the polymer molecule. The physical and dynamic properties under such condition could be further affected due to the molecular confinement in the ultra-thin films. For example, it has been found that the glass transition temperature could be significantly lower or higher than that of the bulk depending on the strength of substrate-polymer interactions [7]. On the other hand, many studies have indicated that, the interactions of the polymer chain with the solid substrate might retard the chain mobility due to the pinning effect [8,9]. Furthermore, Frank et al. [10,11] studied the crystallization behavior of poly(ethylene oxide) (PEO) in the ultra-thin films by using hot stage AFM, grazing angle reflection FT-IR spectroscopy and steady-state fluorescence spectroscopy. They observed that, the degree of crystallinity decreases steadily when the film thickness is less than 200 nm. Taguchi et al. [12,13] found that, for isotactic polystyrene (i-PS), not only the crystallization rate decreased greatly in the ultra-thin film but also the crystal morphology showed great dependence on the film thickness.
The development of atomic force microscopy (AFM) has provided a new capability for determining polymer morphology with nanometer or better spatial resolution. While AFM undoubtedly yields topographic data directly and with superior resolution to conventional optical microscopic methods, it is the characterization of local properties that AFM-based techniques have stimulated widespread interests. A wealth of information about the surface stiffness [14,15], friction [16,17], adhesion [18–20], and thermal properties [21], is readily obtainable. Lateral force microscopy (LFM), which is sometimes referred to as the frictional force microscopy (FFM), could offer insights into friction and wear properties by measuring the lateral force between a sample surface and a sliding probe tip on the micrometer and nanometer scale [22–24]. It has been demonstrated to be a powerful tool to investigate the molecular motions of polymers such as polystyrene (PS) [25–28], poly(methyl methacrylate) (PMMA) [29,30], poly(ethylene terephthalate) (PET) [29,30], poly (vinly alcohol) [29] and poly(vinly acetate) [29,30]. The force-distance curve measurement could also be carried out by AFM, which can provide the detailed information on local elastic and adhesion properties of the sample.
In the present article, we report on the probing of the surface mechanical properties of PVME using AFM. The lateral force and adhesion properties of PVME films and the dependence on the scanning rate as a function of the film thickness are investigated. The correlations among the lateral force, adhesion force and viscoelastic properties of PVME in the confined space of ultra-thin film will be discussed. Furthermore the effects of blending with isotactic polypropylene (i-PP) on the lateral force of the film will also be studied.
2 Experimental
2.1 Materials and sample preparation
The poly(vinyl methylether) (PVME) used in this study was purchased from Scientific Polymer Products, Inc. It has a weight average molecular weight, Mw, of 90 000. The bulk Tg of the PVME is –24 °C, as measured by Modulated DSC 2920 (TA Instruments), which is well below the room temperature. The isotactic polypropylene (i-PP) with Mw = 250,000 was obtained from Aldrich Chemical Company.
Thin PVME homopolymer films were prepared by spin-casting a xylene solution onto a silicon (Si) wafer with a spinning speed of 3000 rpm. Various desired film thicknesses were obtained by varying the weight concentration of the solution in a range from 0.5% to 10%. Prior to the spin casting, the wafers were acid cleaned in a bath of 70/30 volume ratio solution of 96% H2SO4 and 30% H2O2 for 1.5 h at 80 °C, followed by ultrasonic cleaning in acetone, in 2-propanol, and in deionized water. The blend films were prepared by dissolving and mixing i-PP and PVME with different compositions in xylene at 130 °C for 1.5 h to completely dissolve the polypropylene, followed by spin-casting the blend solution onto a silicon wafer at 190 °C. After spin-casting, the film was dried overnight in a vacuum oven in order to evaporate the solvent completely. The thickness of the film was determined using atomic force microscopy (AFM) by scratching the film and averaging line profiles perpendicular to the scratch direction within a rectangular area.
2.2 AFM measurement
The atomic force microscopy measurements were performed on a commercial system (Explore, Thermo Microscopes) in contact mode at room temperature. The cantilever was fabricated from silicon nitride (Si3N4) and designed in a V-shape with a probe tip integrated onto the underside of the cantilever. The spring constant of the cantilever is 0.032 N/m. The radius of the attached tip is about 20 nm according to the manufacturer. The deflection and torsion of the cantilever are measured with a four-segment photo detector using a laser light irradiating the backside of the free end of the cantilever, which are used, respectively, to obtain topographic and lateral force images. The lateral force could be determined from the difference of the histograms of the trace and retrace LFM images. Topographic contributions to the overall lateral force were independent of the scanning direction and thus could be removed by the subtraction process. In the present work, all LFM measurements were under the same loading (10 nA). More explicit descriptions for principles of LFM are referred to the pioneering works [22–26].
Local adhesion properties of PVME homopolymer thin films (thickness less than 150 nm) with different thicknesses were measured by indenting the surface with a probe tip. The cantilever deflection as a function of the displacement of the cantilever (δp) (force–distance curves) during the indentation was recorded. Here, a silicon probe with a high spring constant (42 N/m according to the manufacture) was chosen because of two reasons. Firstly, a high stiffness probe is needed in order to pull the probe out of the film without defection signal saturation due to the high stickiness of PVME film. Secondly, with high stiffness, the probe could indent through the whole film so that the indentation depth could be determined by knowing the thickness of the film. The variance of the indentation depth arising from the sample properties and probing rate changes could be avoided.
3 Results and discussion
3.1 Lateral force of the PVME homopolymer
It has been suggested that the lateral force is dependent on the surface thermal molecular motion. It is related to the energy required to deform the polymer by the cantilever tip, while surface viscoelastic property of the polymeric material influences the contact configuration during the scan of the tip and, therefore, affects the frictional behavior. The lateral force alteration with measuring the temperature and scanning rate corresponds well to the temperature and frequency dependence of the dynamic mechanical properties [25]. The size of the mound formed on a glassy surface is small because of the high elasticity of the surface. It is also small on the rubbery surface with small elasticity because of the fast relaxation of the deformation. The extent of polymer deformation by the tip is expected to be the largest when the sample is in the glass-rubber transition state.
Fig. 1 shows the scanning rate dependence of the lateral force of PVME film with different thicknesses. For the 400 nm-thick film, the lateral force increases with the scanning rate at the low scanning rate region, and then saturates at a high rate region. This result is consistent with the rubbery state of the PVME at room temperature. As the thickness of the film decreases, the lateral force is less dependent on the scanning rate. Especially when the thickness is below 15 nm, the lateral force shows almost no dependence on the scanning rate. The lateral force at the scanning rate of 10 μm/s as a function of the film thickness is depicted in Fig. 2. As could be seen from Fig. 2, the lateral force decreases greatly with decreasing film thickness when the film thickness is less than 200 nm. The lateral force for the 5 nm-thick film is only about one tenth of that for the bulk film. With low loading applied, the probe tip should only indent and characterize a few nanometers of the film surface. Therefore, this low scanning rate dependence and decreasing lateral force in the ultra-thin film should reflect the surface viscoelastic property changes of the polymer film under confinement.
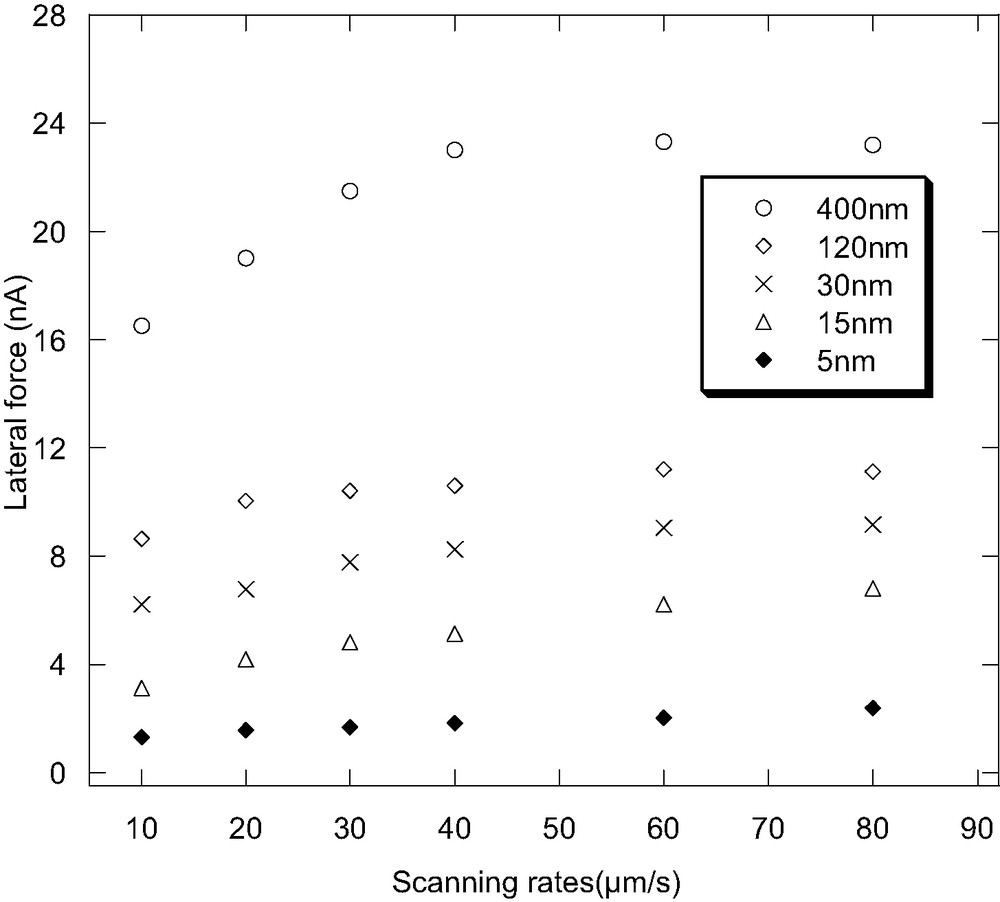
The scanning rate dependence of the lateral force of PVME film with different thickness.
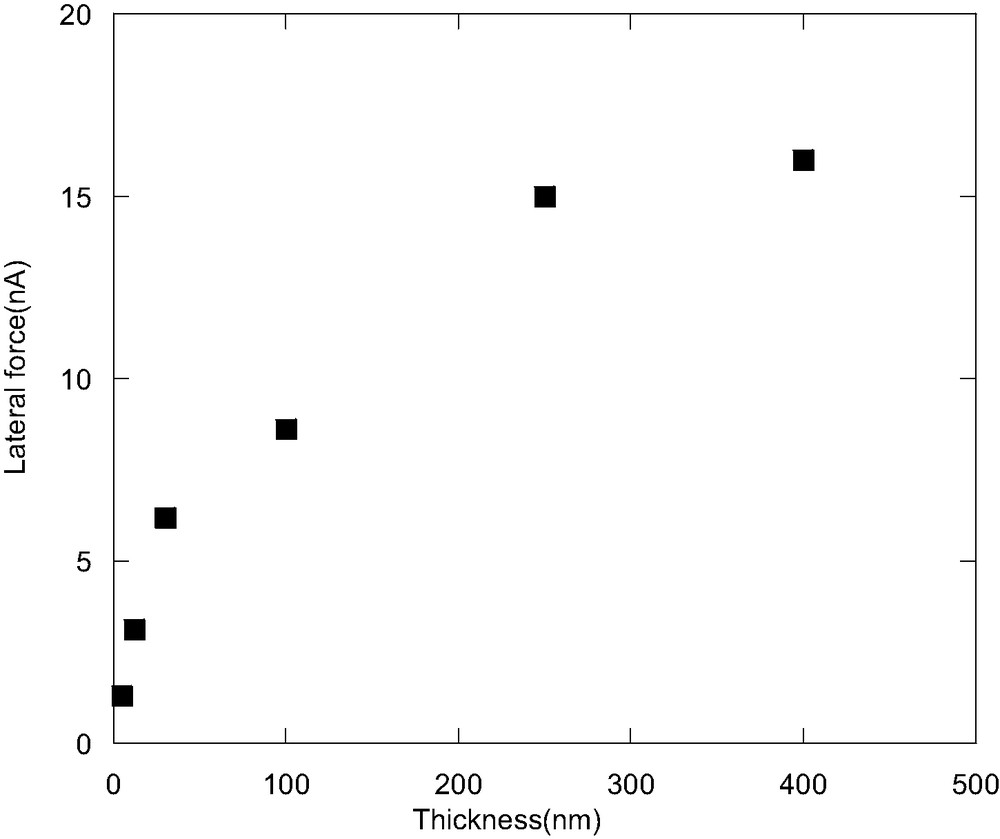
The lateral force as a function of the film thickness at the scanning rate of 10 μm/s.
Recently Granick et al. [31–33] have used the surface force apparatus (SFA) measurement to study the nanorheology of the ultra-thin polymer melts under confinement. It was found that when the film thickness was less than 5-6 times the estimated unperturbed radius of gyration of the polymer chain (Rg), a transition from viscous liquid-like to elastic rubber-like response occurred. The storage shear modulus (G′) increased with diminishing thickness, and the G′ became much higher than the loss shear modulus (G″) meaning that the loss tangent decreased with diminishing thickness. This phenomenon could be interpreted as the similar caging effects. For the bulk polymer melts, this is traditionally referred to as entanglements; polymer chains can slide by one another but cannot cut across one another, and chain motions become highly correlated. Similarly in the ultra-thin films, the motions of the polymer chains might be blocked due to the interaction between the polymer and the substrate such as the pinning effect. It has also been reported by Tabor et al. [34] that there is a relationship between the frictional force under hydrostatic pressure and the dynamic mechanical properties shown by the following equation (1):
| (1) |
3.2 The force–distance curve of PVME homopolymer
The adhesion properties of PVME homopolymer films with different thicknesses are characterized by the force-distance measurements. Fig. 3 shows the force-distance curves for the 20 nm-thick PVME film at probing rates of 0.5 μm/s. It is likely that the fluctuation of the baseline in the curves is due to the optical interference caused by the He-Ne laser. The horizontal axis represents the δp, which is the sum of the deflection of cantilever (δc) and the sample indentation (δs). The consistency of the final curve slope with the spring constant of the cantilever indicates that the probe goes through the whole film, as δp equals δc in such conditions. The adhesion force is defined as the largest pull-off force in order to detach the probe from the sample surface as indicated in Fig. 3.

Force-distance curves for the 20-nm-thick PVME film at probing rates of 0.5 μm/s.
Fig. 4 shows unloading force-distance curves for the 20 nm-thick PVME film at different probing rates. The adhesion forces as a function of the probing rate for the 20 nm and 120 nm thick films are plotted in Fig. 5. In both the 20-nm- and 120-nm-thick films, the adhesion force increases with increasing probing rates at the low rate region. This observation is similar to that of the scanning rate dependence of the lateral force as shown before, although the 20-nm-thick film seems to show less probing rate dependence. It is interesting to note that this probing rate dependence result is opposite from the behavior of poly(tert-butyl acrylate) (PtBuA) results observed by Wang et al. [35]. The difference could be due to the different adhesion failure mechanism. The interfacial adhesion force between the sample and the tip is proportional to the creep compliance (J(t)) [35], which is a function of time and temperature and quite high for PVME at room temperature and low probing rate. Hence, the interfacial adhesion is much higher than the cohesive strength of the sample and the fracture should occur cohesively for the present case. In this case, theories developed for the adhesion of the elastic materials cannot be applied here.
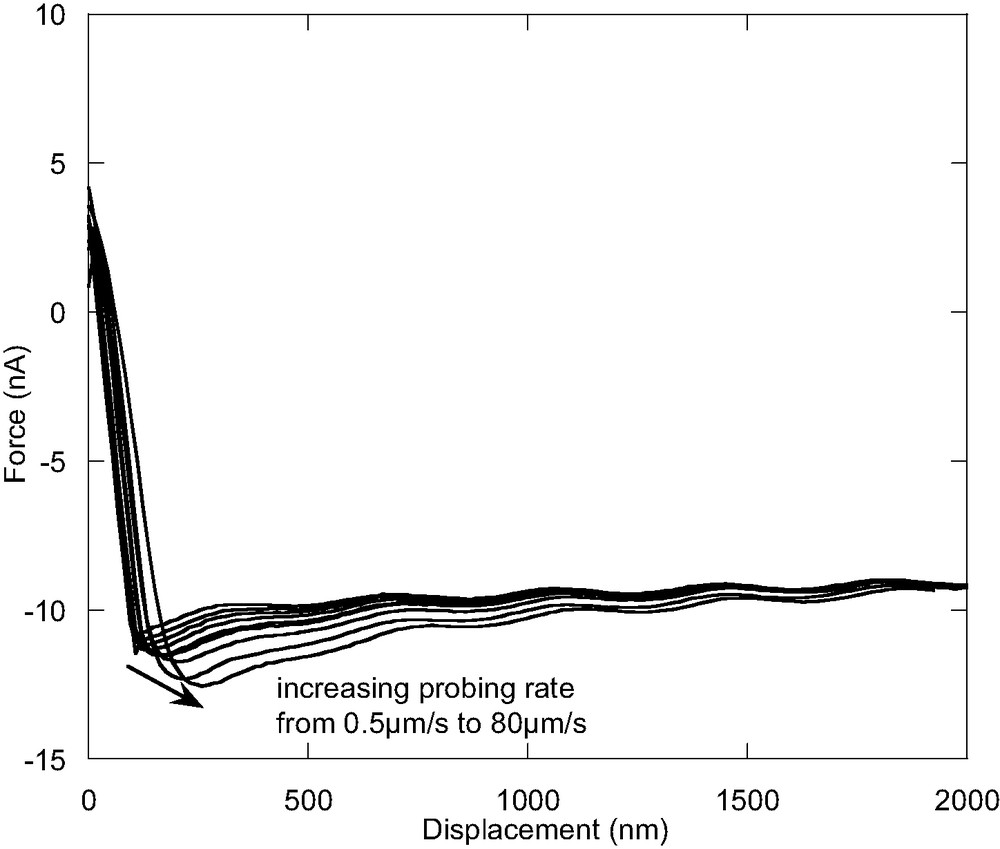
The unloading force–distance curves for the 20-nm-thick PVME film at different probing rates.
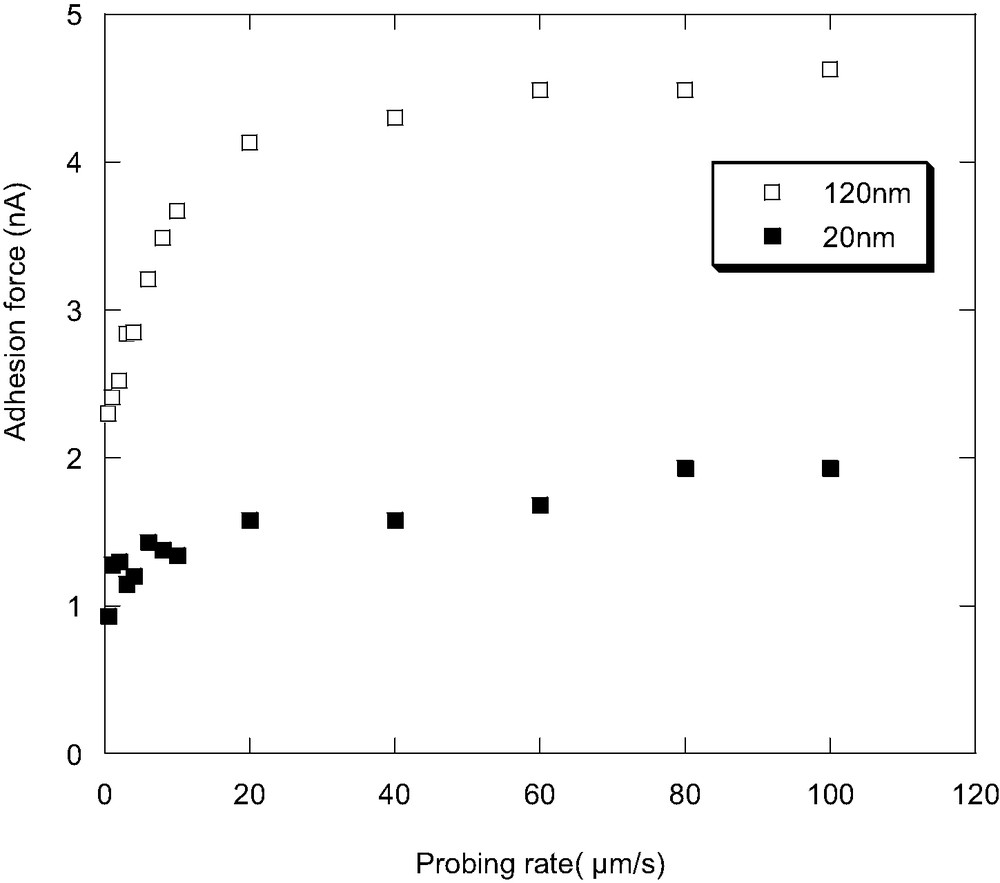
Adhesion forces as a function of the probing rate for the 20-nm-and 120-nm-thick films.
It is interesting to study the adhesion force changes upon decreasing film thickness, which is illustrated in Fig. 6. Only the adhesion force at low probing rates are plotted in order to ensure that the sample shows fluid-like behavior. The indentation deformation is totally plastic in this condition and the adhesion failure occurs cohesively. In this case, it is assumed that the adhesion force is proportional to the contact area between the probe and the sample. The contact area (A) should be proportional to δs1.5 or δs2, depending on the parabolic or the conical geometry of the probe, where δs is the sample indentation depth. In the present study, the maximum sample indentation depth should equal to the thickness of the film. By using the adhesion force of 120-nm-thick film at the rate of 0.5 μm/s as the reference, the adhesion force is proportional to δs2, which is illustrated by the dashed line in Fig. 6. It is obvious from comparing the shape of the dashed line and experimental results that, the adhesion force per unit area increases with the decreasing film thickness if normalized by the contact area.
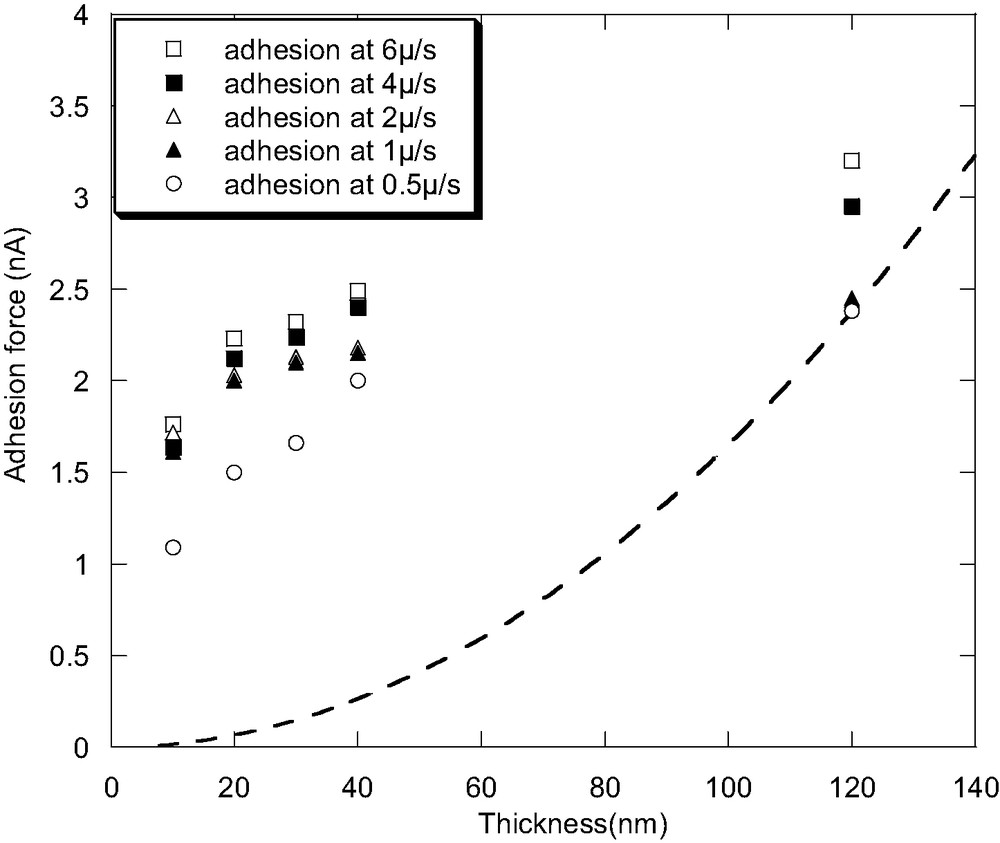
Adhesion force of the PVME thin film as a function of film thickness under different probing rates, the dashed line illustrates the adhesion force proportional to δs2 by using the adhesion force of a 120-nm-thick film at a rate of 0.5 μm/s as the reference.
Although quantitative results should be based on the exact measurement of the contact area and the probe geometry, the present results is interesting in that it is opposite to that for the ultra-thin PtBuA film observed by Wang et al. [35] and also apparently opposite to the lateral force results discussed above. However, such discrepancy could be consistent in reflecting the viscoelastic property changes of the polymers in the ultra-thin films. Wang et al. [35] found that, in PtBuA ultra-thin films (less than 20 nm), the interfacial adhesion between the sample and the tip is greatly suppressed. They interpreted it as the existence of the immobile dead polymer layer in immediate vicinity of the substrate. Therefore, polymer films stiffen in the ultra-thin films, and the interfacial adhesion between the tip and the sample (Fad) decreases as Fad ~ 1/E. On the other hand, in the present study, the adhesion fails cohesively. Hence, it is reasonable to find the increase of adhesion as the stiffness of the sample increases in the PVME ultra-thin films while the lateral force decreases at the same time. In this sense, those results are consistent with each other suggesting the decreased polymer chain mobility in the ultra-thin films reported by the recent literatures [8,9], and the increased modulus and the decreased loss tangent as indicated by the surface force apparatus measurements [31–33]. It should be noted here that the substrate in the ultra-thin film might have great effect on both the chain mobility and the adhesion measurement [7]. Studies are currently on going to clarify this point by comparing results of the same polymer but on different substrate, with well known interfacial interactions with polymer.
3.3 Lateral force measurement of i-PP/PVME blends
It has been found that when i-PP/PVME blend is prepared by casting the solution at 190 °C, it shows a homogeneous morphology [36]. Therefore, it is interesting to study the lateral force of the film upon blending as the polypropylene is semi-crystalline while the PVME is amorphous. The lateral force microscopy has been used to characterize the surface structure of amorphous polymer blends [37]. However, to the authors' knowledge, there is not report using the lateral force microscopy to characterize the surface structure of a semicrystalline polymer blend. Fig. 7 shows the scanning rate dependence of the lateral force of i-PP/PVME blend with different compositions. Firstly, it is interesting to find that, for semi-crystalline polypropylene, the lateral force decreases with increased scanning rate, which is opposite to the scanning rate dependence of PVME homopolymer film. As it has been discussed before, the scanning rate dependence of the lateral force is related to the surface molecular motion or the glass transition. For polypropylene, it is less likely that such scanning rate dependence relates to the molecular motion of the amorphous region of i-PP. As the glass transition of the amorphous phase of i-PP is around 0 °C, which is below room temperature, the lateral force should increase with increasing scanning rate. We, therefore, correlate such scanning rate dependence to disordered crystalline structure of the i-PP surface. Recently, Androsch et al. [38] used temperature-modulated DSC (TMDSC) and AFM to show that when the samples of i-PP is crystallized from the melt at high cooling rates, the polypropylene crystals at the surface would have a portion of reversible melting, which starts from 23 °C till the melting temperature of i-PP. Our hypothesis could be further supported by the fact that the lateral force is greatly decreased after the i-PP sample is annealed at 130 °C for two hours. The lateral force did not decrease but increased with the increasing scanning rate, which might be due to the effect of the i-PP surface amorphous phase. It should also be noted that the lateral force shows little dependence on the scanning rate for the blend with the fraction of PVME from 0.2 to 0.7. With the fraction of PVME around 0.7, the lateral force starts to show a small increase with the scanning rate increase, which is similar to that of PVME homopolymer. For i-PP/PVME blends, as the surface shows a homogenous morphology, it is assumed that both the amorphous and disordered crystalline regions should contribute to the lateral force. Those scanning rate dependence results are reasonable since the scanning rate dependences of lateral force of these two homopolymers at room temperature are opposite. Even with the addition of a small amount of PVME, the scanning rate dependence of the lateral force would not resemble that of i-PP. However, it should be noted that, as the glass transition of PVME increases upon blending and the crystalline structure of i-PP is also greatly disrupted by the addition of PVME as shown by the previous paper [36], these might also affect the scanning rate dependence of the lateral force.
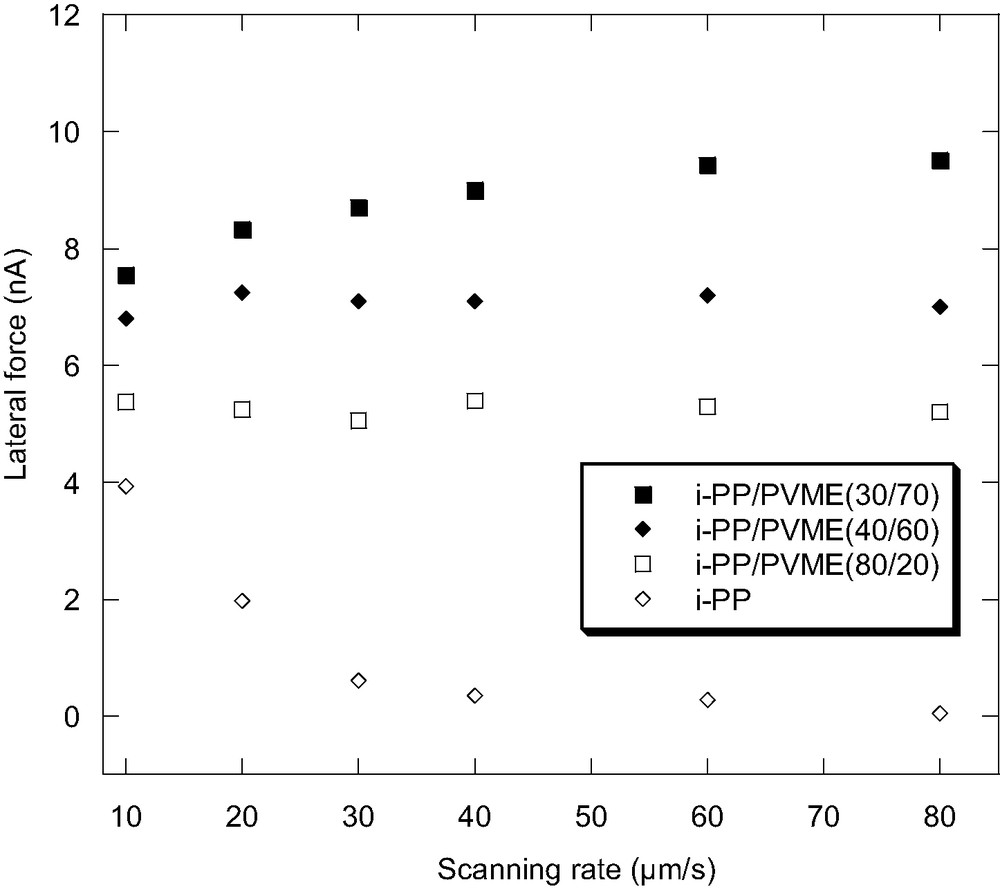
Scanning-rate dependence of the lateral force of i-PP/PVME blends with different compositions.
Fig. 8 shows the lateral force of the blend at the scanning rate of 10 μm/s as a function of PVME composition. As shown in the figure, the lateral force of the i-PP homopolymer film is smaller than that of the PVME film, which can be attributed to the high elastic modulus of the semicrystalline surface structure of i-PP film. As the PVME concentration increases, the lateral force increases slowly at first and shows a maximum when the composition of PVME is around 80%. As stated before, for these amorphous/semi-crystalline blends, both the amorphous and crystalline regions contribute to the lateral force measured. As the lateral force of the amorphous phase is much higher than that of the crystalline phase of i-PP, the lateral force should be attributed mostly to the amorphous region when the PVME concentration is high. On the other hand, upon blending with i-PP, the glass transition of PVME phase increases and is closer to room temperature. In this case, the dissipation of energy is higher since the formation of the rim ahead of the probing tip needs high energy owning to the limited molecular mobility. The relaxation time is not short enough to recover the deformed surface and, hence, the lateral force is higher. Therefore, it is reasonable to find that the lateral force increases with the addition of a small amount of i-PP. However, with the concentration of i-PP further increasing, the fraction of the crystalline region of the blend sample increases and contributes more to the lateral force while the fraction of amorphous region decrease; and therefore, the whole lateral force starts to decrease. With the further addition of i-PP, the amorphous content decreases, and, therefore, the lateral force decreases further. It is interesting to note that similar lateral force peak is also observed when the polymer material goes from the rubbery state to the glassy state. However, in the present study, the peak is not due to the glass transition of the polymer material, but due to the blending of amorphous and semicrystalline polymers.
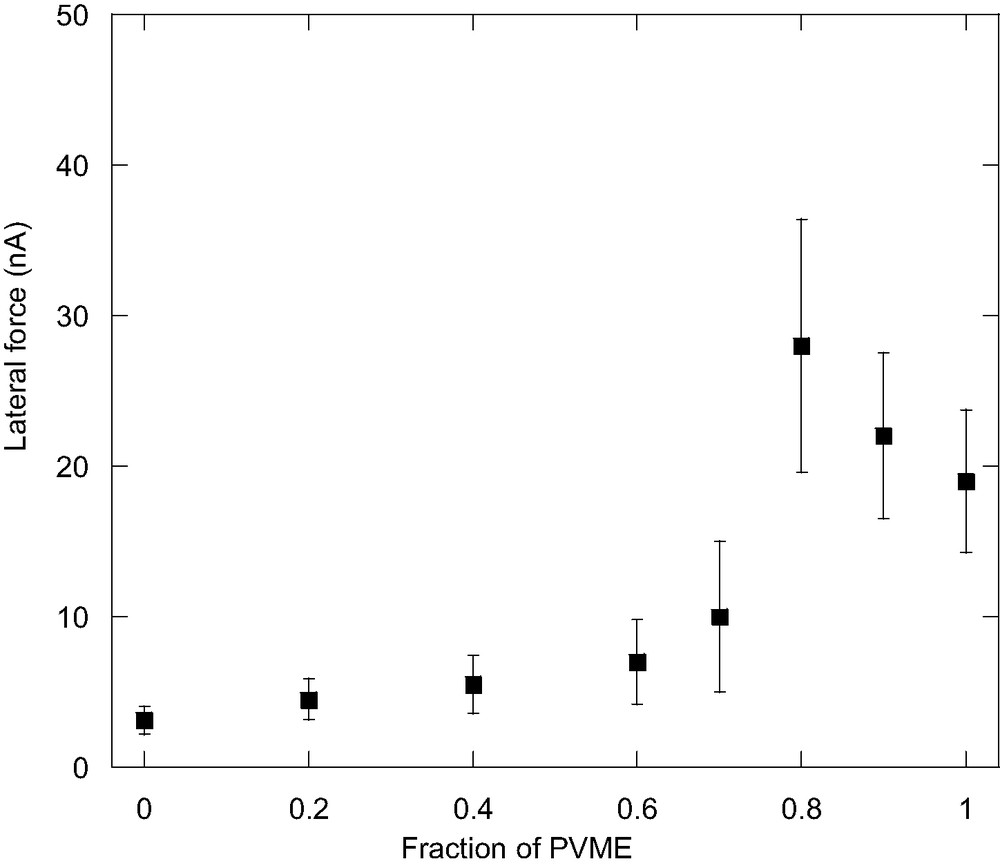
The lateral force of the i-PP/PVME blends as a function of PVME composition at the scanning rate of 10 μm/s.
4 Conclusion
In summary, the atomic force microscopy (AFM) was used in the present article to characterize the surface mechanical properties of the poly(vinyl methylether) (PVME) homopolymer and blends. The effects of the confinement in the ultra-thin film and the blending with semicrystalline isotactic-polypropylene (i-PP) were discussed. When the thickness of the film is less than 200 nm, lateral force decreases with the film thickness decrease and shows less dependence on the scanning rates on the basis of lateral force microscopy (LFM) observations. On the other hand, the adhesion force per unit area increases with the decreasing thickness on the basis of local adhesion measurements. Although the lateral force results and adhesion measurement results are seemingly contradictory, they are consistent in that both reflect the decreasing polymer chain mobility and the increasing polymer stiffness due to the confinement in the ultra-thin films. On the other hand, upon blending with i-PP, both the lateral force scale and its scanning rate dependence have changed. Similar to the behavior when the polymer films go through the glass-transition, the lateral force shows a maximum when the fraction of PVME is around 0.8, which could be interpreted by the contributions and fraction changes from both amorphous and crystalline regions of the blends. The increase of the lateral force with the addition of i-PP at low i-PP composition region is due to the increasing glass transition of the film. And the decrease of the lateral force with the high i-PP composition regions is due to the decreasing amorphous fraction of the film surface.
Acknowledgments
The authors gratefully acknowledge the financial support of BP-Amoco Corporation.