

1 Introduction
The electrochemical and photophysical properties of fullerene C60 (including its derivatives and higher homologues) make it an ideal candidate as an electron/energy acceptor in supramolecular assemblies. Indeed, the discovery of buckminsterfullerene C60 coincided somewhat fortuitously with the very beginnings of the area of supramolecular chemistry itself [1]. The fields of fullerene and supramolecular chemistry have since matured and overlapped considerably and today the literature displays many wonderful examples where fullerene C60 have been employed in such assemblies. An obvious question, and one which has preoccupied chemists interested in harnessing the potential of fullerene C60, is what are the most appropriate electron/energy donors to be used in parallel with C60. There are many criteria which must be met which include reasonably long lived excited state lifetimes, good absorption in the UV/Vis and good photostability. Many candidates are possible ranging from porphyrins [2–5] and phthalocyanines [5,6] to conjugated organic oligomers [7,8]. In this account we will focus on the use of transition metal complexes in the role of electron/energy donors when excited on their long-lived metal-to-ligand-charge-transfer (MLCT) excited states. In particular we will present fullerene hybrid systems comprising tris(2,2′-bipyridine)ruthenium(II), (2,2′-bipyridine)rhenium(I) or bis(1,10-phenanthroline)copper(I) subunits. The MLCT excited states of such moieties have a marked reducing character that, in principle, make them ideal partners for the construction of donor–acceptor systems with C60 fullerenes.
We present here three systems of varying degrees of sophistication that we have investigated; a relatively simple C60-metal complex donor-acceptor dyad, an elegant metal-complexed rotaxane containing fullerene C60 stoppers and finally a series of C60 functionalized dendrimers with metallic cores.
2 Dyads with Re(I)- and Ru(II)-complexed moieties
The photodynamic processes of dyads F–Re and F–Ru (Scheme 1), where a methanofullerene unit is linked to a Re(I) and Ru(II) bipyridine-type complex were investigated in CH2Cl2 solution [9].
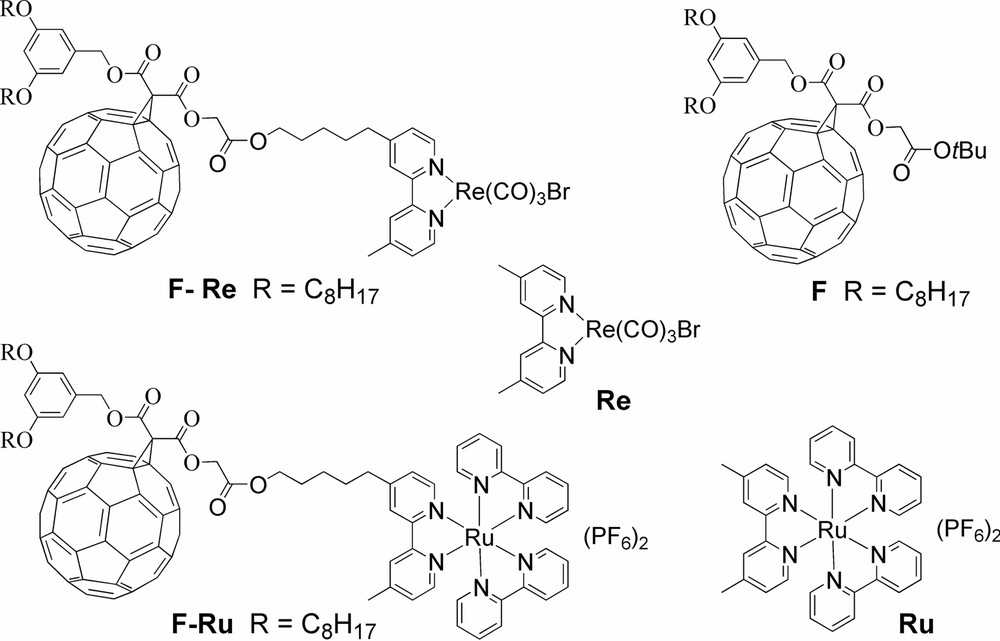
Structures of dyads F–Re, F–Ru, and their model compounds.
Electrochemical studies were carried out using cyclic voltammetry and showed marked differences between these two systems. In the case of F–Re, fullerene centered reductions and metal centered oxidations appear shifted to more positive potentials when compared to the model compounds F and Re. This is not observed in the case of F–Ru, its oxidation and reduction potentials being substantially unchanged when compared to F and Ru. These results indicate the presence of small electronic interactions between F and Re in F–Re, and the absence of such interactions between F and Ru in F–Ru. We believe this is because the dyads adopt different conformations in solution, with F–Re having preferentially a folded conformation and F–Ru an extended conformation. This can be rationalized by considering the overall charge of the two metal complexes. The ruthenium complex in F–Ru bears a double positive charge which will repel the hydrophobic fullerene unit, whereas the rhenium moiety of F–Re is neutral and there is no such repulsion between the fullerene and rhenium units. F–Re can then adopt a folded conformation where F and Re are close, thus affecting electrochemical potentials. It should of course be pointed out that the different conformations are possible due to the flexible alkyl linker connecting the fullerene and metal complexes in these dyads. Such electrochemical trends have been noted in similar systems [10].
The absorption spectra of F–Re and F–Ru in CH2Cl2 are depicted in Fig. 1 and match the profiles obtained by summing the spectra of the component units within an experimental uncertainty of ± 15% [9]. For F–Re, the absorption features of the C60 unit largely dominate the entire spectral profile, unlike F–Ru. Importantly, in the case of F–Ru an almost selective excitation of the metal-complexed moiety ( ≥ 85%) is possible at 456 nm, corresponding to the maximum of the MLCT absorption band. At λ > 600 nm, the C60 moiety can be excited exclusively. In contrast, selective excitation of the Re(I) moiety is not possible in F–Re and the highest extent of excitation of such a unit is achieved at 292 and 380 nm, that is 35 and 30%, respectively. Conversely, selective excitation of the C60 moiety is possible at any wavelength above 490 nm.
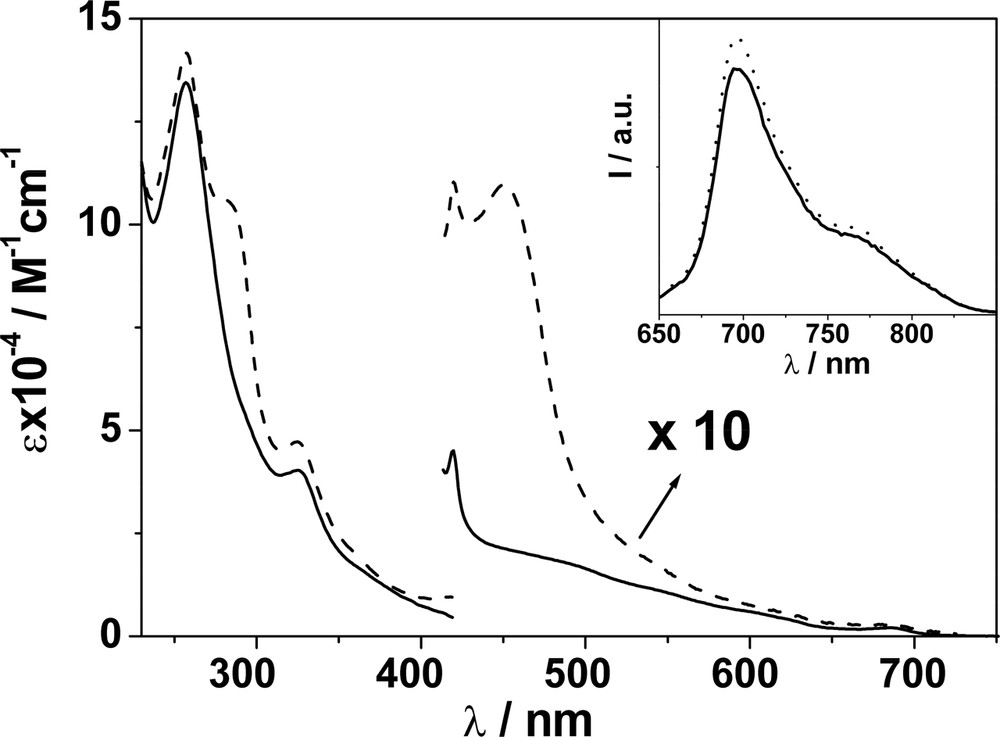
Absorption spectrum of F–Re (__) and F–Ru (ـ ـ ـ) in CH2Cl2; beyond 420 nm, spectra are increased by a factor of 10 for display purposes. The inset shows fluorescence spectra of isoabsorbing solutions of F (···) and F–Re (__); λex = 550 nm, where only the C60 moiety absorbs in F–Re. Identical fluorescence relative intensities are obtained by comparing the spectra of F and F–Ru upon excitation at 625 nm.
F exhibits the typical short-lived methanofullerene fluorescence band at ~ 700 nm (inset Fig. 1) [11], that is also observable at 77 K. The latter result allows the location of the lowest singlet fullerene excited state at 1.78 eV. We take the energy value of the lowest fullerene triplet excited state to be 1.50 eV, as estimated for methanofullerenes by theoretical calculations [11].
Re and Ru exhibit typically intense and long-lived MLCT luminescence bands at 298 and 77 K (Fig. 2). When F–Re and F–Ru are excited (not selectively, see above) at the metal complex moiety at 298 K, the strong and long lived MLCT luminescence of both the Re and Ru units are quenched significantly (left hand side panels in Fig. 2). The quenching rate constants were evaluated from experimental data and are practically identical; 2.5 × 108 s–1 (F–Re) and 2.3 × 108 s–1 (F–Ru). Fullerene fluorescence is also observed in both systems but is not, however, the result of C60 singlet sensitization from the 3MLCT excited state of the Re(I) and Ru(II) centers. Rather, the measured quantum yield of C60 fluorescence reflects the amount of light directly exciting the fullerene moiety. Upon excitation at 550 nm (F–Re) and 625 nm (F–Ru), where only the C60 chromophore absorbs, the typical methanofullerene fluorescence band is substantially unaffected for both dyads if compared to that of the reference compound F, as the inset in Fig. 1 shows for F–Re. These results indicate that intercomponent photoinduced processes do not occur when the C60 chromophore is excited, rather, only when the metal complex is excited directly.
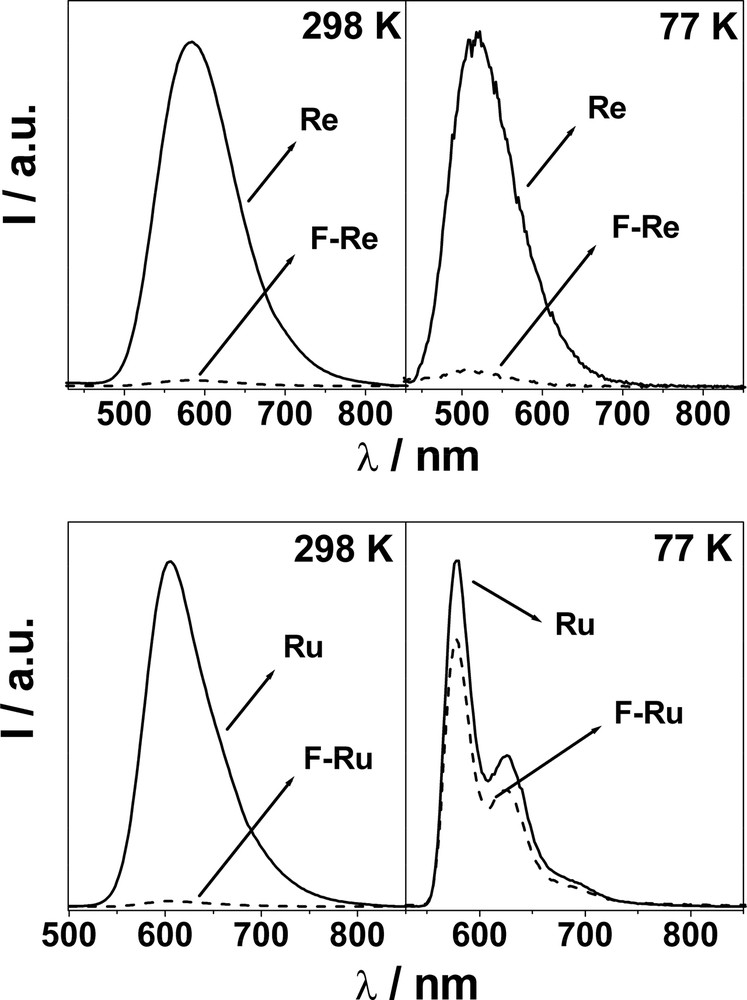
Left-hand side: 3MLCT luminescence spectra of Re and F–Re, and Ru and F–Ru at 298 K in CH2Cl2. Right-hand side: 3MLCT luminescence spectra of Re and F–Re and Ru and F–Ru at 77 K in CH2Cl2/MeOH (1:1 v/v). (isoabsorbing solutions, λex = 292 nm for Re and F–Re; λex = 456 nm for Ru and F–Ru).
On the basis of the energy of the lowest excited states of the various moieties, as determined by emission spectroscopy, we can construct an energy level diagram for the two dyads (Fig. 3). The electrochemical results allow the location of a charge-separated state (CS) corresponding to the energy difference between the reduction of the C60 moiety and the oxidation of the metal-complexed unit [F––M+] [12]. Owing to its energy position (≈1.95 eV in both cases), such a CS state might play an active role in the cascade of photoinduced processes following 3MLCT excitation. The presence of fullerene triplet (3F–M) was confirmed in these systems by monitoring sensitization of singlet oxygen at 1270 nm.
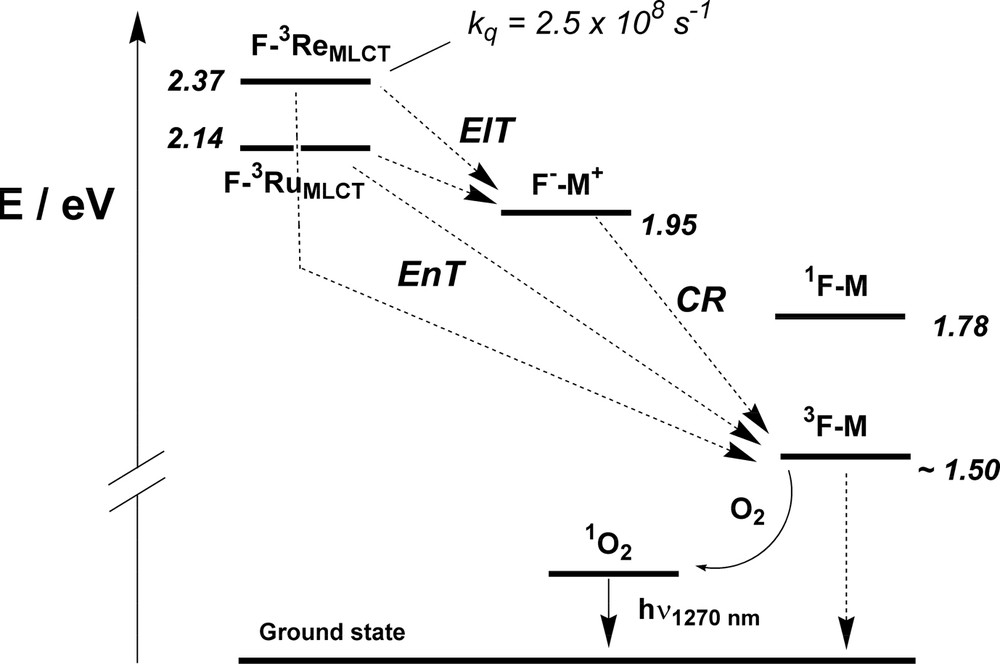
Energy-level diagram showing intercomponent energy- and electron-transfer processes in F–Ru and F–Re. The charge separated level is at the same energy for both dyads.
From the energy level diagram it is evident that, in principle, two different processes can account for the 3MLCT luminescence quenching and the observation of fullerene triplet in these dyads: 1) direct triplet-triplet (T–T) energy transfer (EnT) from the 3MLCT of the metal-complexed unit to the fullerene triplet, or 2) electron transfer (ElT) leading to [F––M+] followed by charge recombination to the fullerene triplet. Energy transfer to the lowest fullerene singlet excited states is ruled out because luminescence data do not show any fullerene fluorescence sensitization. Earlier studies suggest that T–T (Dexter-type) EnT processes display strong similarities with electron transfer processes and can be treated by a similar formalism [13,14]. In the case of dyads made of a Re(I) complex and an aromatic subunit the rate constant of T–T EnT as a function of reaction thermodynamics obeys a Marcus-type behavior, where the maximum rate is obtained for –ΔG0 ≈ 0.3–0.4 eV [15]. In our case –ΔG0 = 0.87 (F–Re) and 0.64 eV (F–Ru), so it is reasonable to assume that the T-T EnT process is positioned in the inverted region, whereas the lower exergonicity of the competing CS processes (–ΔGCS0 in the range 0.2–0.4 eV) is optimal to locate ElT in the proximity of the maximum of the Marcus parabola as has been found, for instance, in ZnII–porphyrins/C60 molecular dyads [16]. The same thermodynamic arguments apply to the CR process which is likely to be faster in giving the C60 triplet (–ΔG0 ≈ 0.55 eV) rather than the ground state (–ΔGCR0 ≈ 1.95 eV). Therefore, on the basis of the above considerations, we would tend to ascribe the quenching of the MLCT excited states to electron transfer rather than to direct triplet–triplet energy transfer.
To find support to the ElT mechanism, we recorded 77 K luminescence spectra for F–Re and F–Ru (right-hand side panels in Fig. 2). It is known that the energy of a charge-separated state is expected to increase in a rigid matrix, where the effective solvent reorganization energy increases dramatically because the solvent molecules cannot reorient to stabilize the newly formed ions [17]. Hence, when the ElT process is slightly exergonic at room temperature, it may become endergonic (blocked) at 77 K. Consequently, for multicomponent arrays where luminescence quenching is observed in solution, the lack of such quenching in a rigid matrix is taken as evidence to support the ElT mechanism in the fluid medium. Very interestingly, the 3MLCT emission of the metal-complexed moiety is recovered for F–Ru and a lifetime of 6.8 μs is measured, which is identical within experimental uncertainties to that of Ru (7.3 μs). This suggests that triplet-triplet energy transfer, though thermodynamically allowed, is extremely inefficient at 77K and is not able to compete with intrinsic deactivation of the Ru moiety, which gives rise to its typical 3MLCT emission. Therefore, at 77 K neither energy or electron transfer occurs in F–Ru. Importantly, since the rates of energy transfer processes are known to be scarcely affected by temperature [18], we can also argue that at 298 K, EnT cannot compete with ElT as the quenching process.
For F–Re, recovery of the 3MLCT luminescence is not observed at 77 K, and a quenched lifetime of 3.7 ns is determined (to be compared to 5.3 μs for Re). We believe that this can be explained considering the higher energy of the 3MLCT level, if compared to F–Ru. A 0.42-eV difference between 3MLCT and the CS state (Fig. 3) is likely to be too large to make the electron transfer reaction endergonic at 77 K, as it happens for F–Ru (–ΔG0 = 0.19 eV). Accordingly, the 3MLCT excited state of F–Re is quenched by electron transfer even in a 77 K rigid matrix, also because the EnT mechanism is likely to be as inefficient as in the case of F–Ru, given the similarity of the involved thermodynamic parameters (Fig. 3).
3 A Cu(I) complexed rotaxane with fullerene C60 stoppers
Cu(I)–bisphenanthroline complexes are excellent prospective partners of fullerene C60 in supramolecular assemblies [19]. This is due to a number of reasons, the first of which is that such complexes are known to be potent reductants in the excited state [19–23]. In addition, when the phenanthroline ligands are substituted in the 2,9 positions by aryl residues, the complexes display excellent photostability, long-lived luminescent excited states and absorption spectra throughout the UV/Vis spectral region [23,24], which make them potentially useful for harvesting solar energy. Finally, it should be pointed out that the outstanding affinity of Cu(I) towards phenanthroline-type ligands allows the high-yield preparation of complex supramolecular structures such as catenanes, rotaxanes, knots and helicates [23,25–28].
We investigated the rotaxane F–Cu formed from a bisphenanthroline Cu(I) complex and two C60-type units which act as stoppers (Scheme 2). The photophysical properties of the rotaxane are compared with those of two suitable molecular models: a C60 methanofullerene (F) [29–31] and a Cu(I) catenate (Cu) [32].
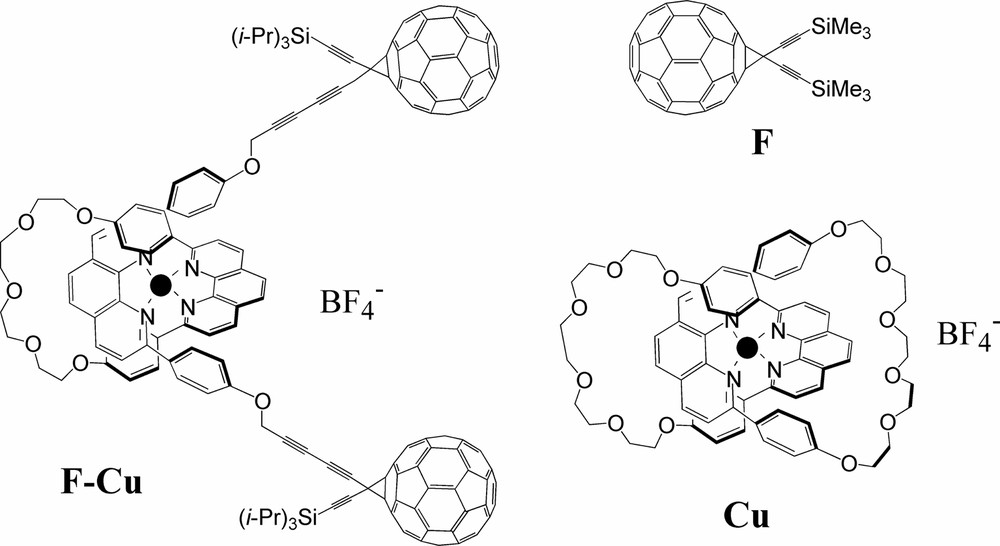
Structures of compound rotaxane F–Cu and its model compounds F and Cu (● = Cu+).
The redox properties of F–Cu were studied by cyclic voltammetry. Surprisingly, a significant anodic shift (~ 300 mV) was observed for the reversible Cu-centered redox process in comparison with other similar mononuclear complexes (including compound Cu) [33]. By contrast, the first reduction of both F and the fullerene units of F–Cu appear in the range typical of C60 monoadducts [34,35].
The electronic absorption and luminescence spectra of F–Cu, F and Cu in CH2Cl2 are shown in Fig. 4. Luminescence studies of F and Cu reveal that they display bands with maxima which are almost coincident at 700 nm. For F–Cu only, a very weak emission was detected, whose shape is similar to that of F. The luminescence lifetime for F was measured to be 1.6 ns, while F–Cu displays biexponential kinetics, with lifetimes of 0.5 and 1.7 ns. The former value is obtained as an average of luminescence (probed at 700 nm) and transient absorption (probed at 520 nm) decays which are in excellent agreement. The picosecond transient absorption spectrum of F–Cu displayed bands with maxima at 520 and 900 nm (also observed for F) corresponding to fullerene singlet and triplet bands, respectively. The triplet appears to be formed with a rate constant (1.8 ns) in agreement with the longer luminescence lifetime.
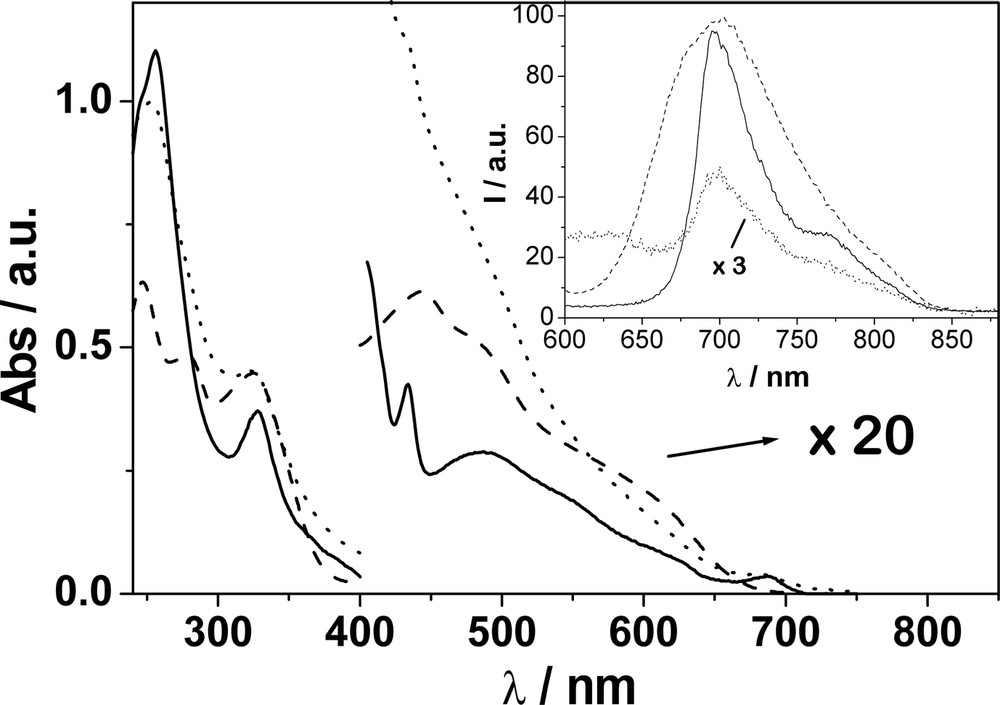
Electronic absorption spectra of F (__), Cu (- - -) and F–Cu (···) in CH2Cl2. In the region 400–800 nm region the spectra are multiplied by a factor of 20 for display purposes. The inset shows the luminescence spectra of isoabsorbing solutions of F (__), Cu (ـ ـ ـ) and F–Cu (…) in CH2Cl2 also. At the excitation wavelength (532 nm) the light repartition is 40% : Cu and 60% : F.
The photophysical properties of the reference compound Cu and related compounds have been extensively investigated [25,36–41]. However, such properties for Cu, as well as for F, appear strongly quenched in F–Cu. In particular we observe: (a) quenching of the fullerene fluorescence; (b) quenching of the Cu MLCT emission and (c) reduction in the yield and lifetime of the fullerene triplet. Such observations clearly indicate the presence of novel competitive deactivation pathways in the excited states of F–Cu with respect to the molecular components.
On the basis of the steady state and time resolved absorption and luminescence data, as well as electrochemical potentials, [11] we can construct an energy level diagram for F–Cu, in the same way as for the dyads in the previous section. Excitation of F–Cu (355 or 532 nm) leads to the population both of the fullerene-centered singlet excited state 1F–Cu and of the F–*Cu state in a 3:2 ratio, according to the absorption coefficients. From the quenched lifetime of the fullerene singlet (0.5 ns, see above), a rate constant of 1.6 × 109 s–1 is derived for the step deactivating 1F–Cu in the rotaxane. Such deactivation was attributed in part to electron exchange (Dexter-type) energy transfer and partly to intersystem crossing to 1F–Cu (steps 1 and 2 in Fig. 5).
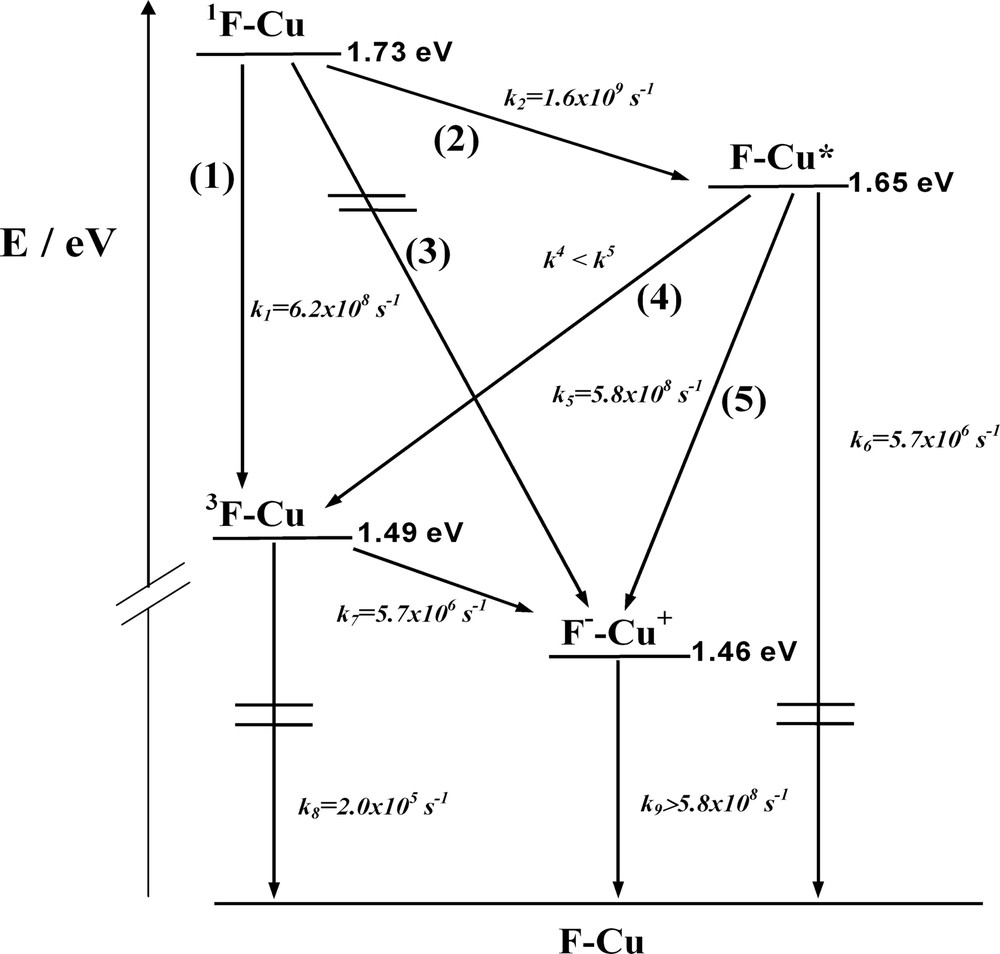
Energy-level diagram describing the intercomponent energy- and electron-transfer processes which occur in the rotaxane F–Cu in CH2Cl2.
The Cu(I)-complexed unit lifetime is quenched from 175 to 1.7 ns, corresponding to a rate constant of 5.8 × 108 s–1. The possible deactivating pathways for the MLCT manifold are: (i) energy transfer to the fullerene localized triplet (step 4) and (ii) electron transfer to the charge-separated state (step 5). The transient absorption data concerning the evolution of the fullerene triplet 3F–Cu indicates that only a negligible part of the observed triplet is formed by the process displaying the lifetime of 1.8 ns, which is in close agreement with the second luminescence decay with a 1.7-ns lifetime monitored for F–*Cu. Since under our experimental conditions 40% of the absorbed photons produces the Cu(I)-complex-centered MLCT excited state directly, we would expect a larger contribution of fullerene triplet formation than actually observed if step 4 were efficient. Therefore, we would tend to ascribe the rate constant of deactivation of the MLCT levels (k5 = 5. 8 × 108 s–1) mainly to electron transfer leading to the charge-separated state (step 5). It should be noted that in step 5 the less favorable ΔG0 for electron transfer (–ΔG0 = 0.19 eV) with respect to step 3 (–ΔG0 = 0.27 eV) can be counterbalanced by the fact that the electron has to be moved over a shorter distance. In fact in both cases it has to migrate to a fullerene unit, but starting from the central metal in step 3 and from the phenanthroline ligand in step 5 (in MLCT excited states, the electron is localized on the ligand) [42]. Finally, the triplet is also quenched, as we can confirm from the lifetime of this state, which changes from 5 μs in F to 170 ns in F–Cu. The quenching of the fullerene-centered triplet (k7 = 5.7 × 106 s–1) can only be assigned to the charge separation step 7, which is a slightly exergonic pathway (–ΔG0 ~ 0.03 eV). Clear evidence of F––Cu+ state formation and charge recombination rates cannot be derived from our transient absorption spectra as the oxidized transient species [Cu(phen)2]2+ and C60– both display very weak absorption bands [43,44].
4 Fullerene C60 dendrimers with a Cu(I) bis-phenanthroline core
Due to their fascinating structures and properties, dendrimers [45–54], including fullerene dendrimers [55–59], have attracted considerable attention in the past decade. A few years ago we reported an investigation on a series of dendrimers with a bis(1,10-phenanthroline)copper(I) ([Cu(phen)2]+) core and peripheral fullerene π chromophores [60] and showed how the surrounding fullerene-functionalized dendritic branches are able to isolate the central Cu(I) complex. The dendrimers and the parent compounds are depicted in Scheme 3.
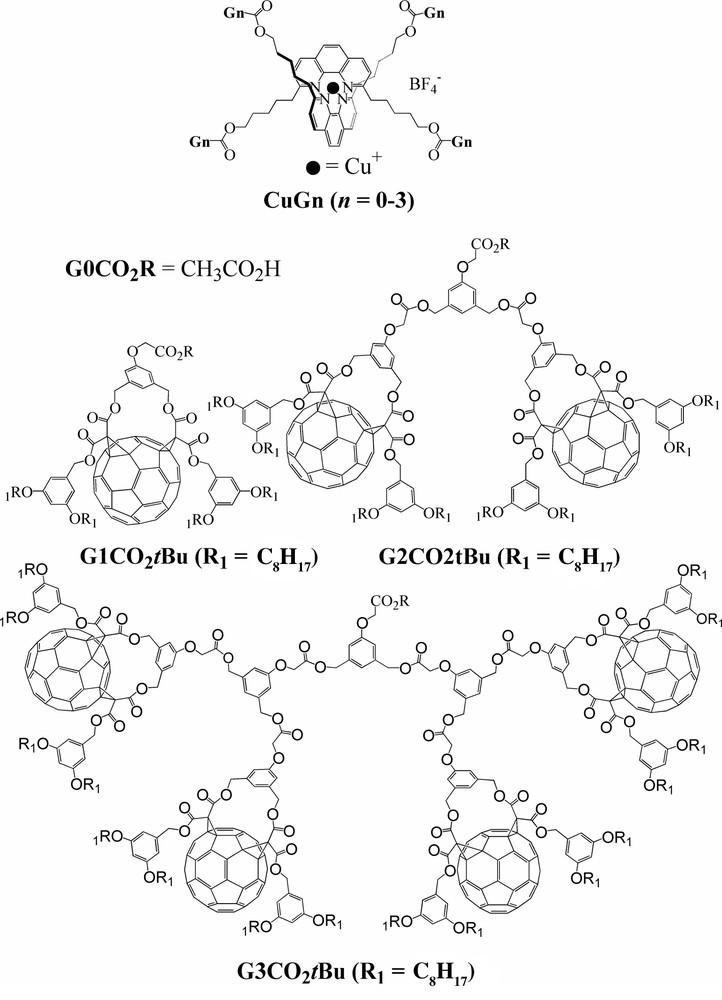
Schematic structures of CuGn dendrimers (top) and related dendrons (bottom).
The redox properties of dendrons GnCO2tBu and dendrimers CuGn were studied by steady-state and cyclic voltammetry in CH2Cl2. G1CO2tBu showed the characteristic behavior previously reported for fullerene cis-2 bis-adducts [61–63]. Similar results that were obtained for reductions of the dendrimers of higher generations G2CO2tBu, G3CO2tBu, and CuG1–CuG3 show that the peripheral fullerene subunits behave as independent redox centers in all compounds. The Cu(I)/Cu(II) oxidation potential of CuG1 remains unchanged relative to the parent compound CuG0, and this suggests the absence of ground-state intramolecular interaction between the central copper(I) complex and the four surrounding fullerene units in CuG1. The amplitude of the fullerene-centered reduction wave is expected to be four times larger than that of the Cu-centered oxidation. Surprisingly, however, the amplitude of the oxidation peak of CuG1 is smaller than expected [64]. In addition, the metal-centered oxidation became irreversible, which indicates a decrease in the electron transfer rate, as was previously observed for other electroactive cores in dendrimers [45,51–53]. This suggests that the bulky fullerene units around the Cu center partially prevent its approach to the electrode surface and, as a result, oxidation at the central core could not be completed on the timescale of the CV measurement. Consistent with this, the electrochemical oxidation of the Cu site could no longer be observed with dendrimers of the highest generations CuG2 and CuG3. The central electroactive site appears to be totally inaccessible owing to isolation by the bulkier surrounding dendrimer structures.
The absorption spectra in CH2Cl2 of CuG0, G1CO2tBu, and CuG1 are shown in Fig. 6. The absorption spectrum of CuG0 exhibits the intense π–π* ligand-centered bands in the UV and the much weaker metal-to-ligand charge-transfer (MLCT) bands in the visible region which are typical of the [Cu(phen)2]+-type chromophore. The absorption spectrum of CuG1 corresponds to the sum of the corresponding component units CuG0 and G1CO2tBu, again indicating negligible ground-state electronic interactions between these two components. The absorption spectra of G2CO2tBu and G3CO2tBu have the same shape as that of G1CO2tBu, but the molar extinction coefficients are respectively two and four times larger than those of G1CO2tBu, indicating that also in these dendrimers there are negligible electronic interactions in the ground state. The inset in Fig. 6 shows luminescence of CuG0, G1CO2tBu and CuG1. Interestingly, luminescence of the dendrimer CuG1 is similar in shape to that of the fullerene rather than the Cu–phenanthroline unit.
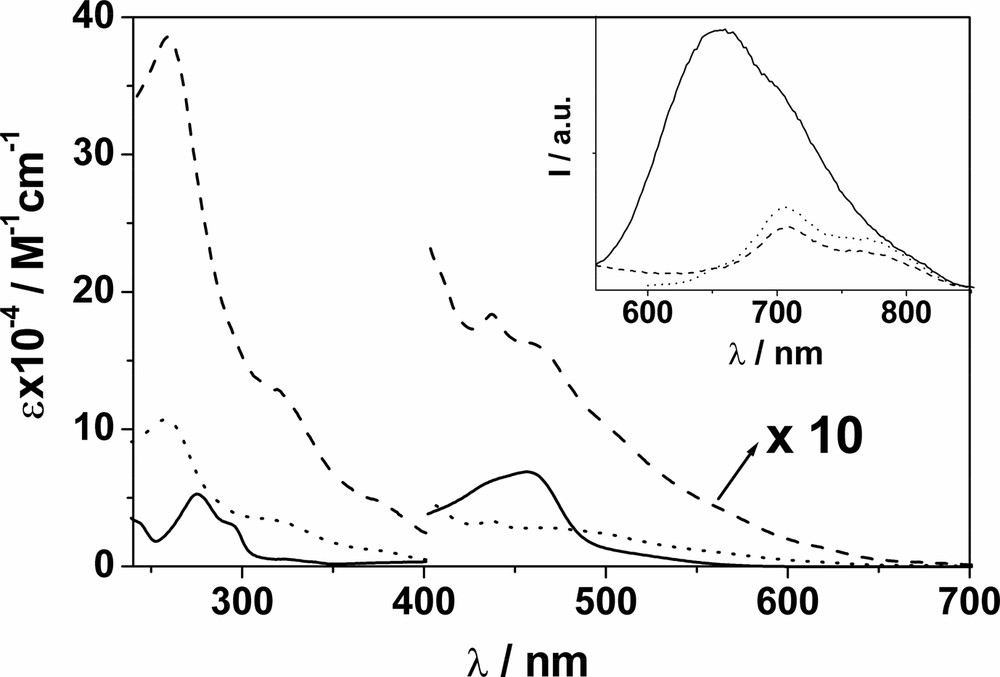
Absorption spectra of CuG0 (__), G1CO2tBu (···), and CuG1 (ـ ـ ـ). Inset: emission spectra of the same compounds at λex = 456 nm (CH2Cl2, 298 K).
Upon selective excitation of the peripheral units at 600 nm, fullerene fluorescence is observed for all dendrimers CuGn; the corresponding emission quantum yield and excited state lifetimes are identical, within experimental error, to those of the corresponding dendronic subunits Gn. Despite the fact that selective excitation of the [Cu(phen)2]+ central core is not possible, clear evidence for the quenching of the luminescence of such a moiety can be obtained. For instance, by exciting CuG1 at 456 nm, although the light partition is about 40% (core) and 60% (fullerenes), the MLCT emission of the central core is dramatically quenched in a steady-state experiment (inset Fig. 6). Analogous patterns are shown by CuG2 and CuG3, although the light partitioning is less favorable for the central core because of the above-mentioned shielding effect. In a time-resolved experiment we did not observe any residual luminescence decay for CuG1 within the time resolution (20 ns); hence, a rate constant of kq > 5 × 107 s–1 can be estimated for the quenching of the luminescence of the central core by the peripheral units. We tentatively attributed the quenching of the MLCT excited state of the Cu(I)-complexed moiety to energy transfer to the C60 singlet or triplet states, even though electron transfer cannot be completely ruled out. [60] Unfortunately detailed transient absorption studies on the fullerodendrimers CuG1–CuG3 upon selective or prevalent excitation of the metal complexed core is impossible due to the unfavorable light partitioning with the overwhelmingly absorbing fullerene moieties.
By comparing the results of the light induced processes in the rotaxane of the previous section and the above described dendrimers an interesting result is that the fullerene moiety is quenched by the Cu(I) center in the rotaxane but not in the dendrimers. The former contains methanofullerenes fragments, the latter bismethano derivatives. In more recent studies involving fullerohelicates made of an identical central dinuclear Cu(I) complex and two external mono- or bismethano derivatives this trend has been confirmed [23,28]. In other words, after having investigated Cu(I)-phenanthroline/C60 arrays for several years having topologies ranging from rotaxanes [11], to dendrimers [60], to sandwich-type triads [23,28] the fullerene moieties are invariably quenched by the metal-complexed unit when methanofullerenes are involved whereas quenching does not occur for bismethanofullerene arrays. This has to do with the inherently different electronic structure of the two fullerene derivatives. By means of an analysis of their fluorescence spectra, which are substantially different, it was possible to conclude that the singlet excited state of methanofullerenes is more prone to undergo electron transfer than that of bismethanofullerenes, thanks to the associated smaller internal reorganization energy [65]. In addition, methanofullerenes are slightly easier to reduce than bismethanofullerenes, giving also a thermodynamic advantage for electron transfer in multicomponent arrays containing the monofunctionalized derivative. The combined effect of these two factors (kinetic and thermodynamic) can explain the different and somehow unexpected trend in photoprocesses of multicomponent arrays containing Cu(I)–phenanthrolines linked to methanofullerenes vs. bismethanofullerenes, which has been found in a variety of molecular architectures.
5 Conclusions
In this account we have presented some of our recent work investigating the photophysical properties of novel supramolecular arrays containing fullerene C60 and transition metal complexes of Ru(II), Re(I), and Cu(I) in CH2Cl2, a solvent of intermediate polarity which allows solubilization of these hybrid architectures. Upon excitation of the metal complexed moieties electron transfer is active in practically all cases. For Ru(II) and Re(I) complexes, the charge separated state corresponding to the oxidation of the metal center and reduction of the carbon cage is located well above the fullerene triplet level. Hence electron transfer is followed by charge recombination to the fullerene triplet state, which act as a sink for the deactivation of upper electronic levels. In the case of the Cu(I)-type hybrids, the CS state may lie lower than the fullerene triplet (i.e. below ca. 1.5 eV), thanks to the strongest reducing character of the MLCT excited state of Cu(I)–phenanthrolines, compared to Ru(II) or Re(I) complexes. Accordingly Cu(I)–phenanthrolines, appear to be more promising candidates with respect to Re(I) or Ru(II)–bipyridines for the construction of hybrid architectures with fullerenes featuring long-lived CS states [66]. However, further efforts are needed in order to elongate the lifetime of the electron transfer pair, which has turned out to be extremely short in our systems investigated so far. Our ongoing research work is aimed at this goal.
Acknowledgements
We gratefully acknowledge all of our colleagues and co-workers who have contributed to this work, their names are cited in the references. We thank the financial support of the European Commission through the RTN Contract ‘FAMOUS’ (HPRN-CT-2002-00171).