

1 Introduction
Photoactive systems comprised of an electron-donor and an electron-acceptor, are particularly attractive for conversion of light into electric current [1–5]. In such systems, rapid photoinduced electron transfer (PET) should be followed by diffusional splitting of a charge separated radical pair, mimicking a key step in natural photosynthesis. The inherent properties of C60, which can accept reversibly up to six electrons [6,7] together with its low reorganization energy [8–10], offer to this field an exceptional three-dimensional electron-acceptor. On the other hand, macrocycles such as porphyrins, subphtalocyanines and their metallated homologues, can be used as electron donors [3,4]. The advantage of these heterocycles lies in the minimal structural changes when electrons are released from them.
The nature of the linker between the two electroactive units controls the distance, orientation and electronic coupling and thus, small structural variations can affect the lifetime of the charge-separated state. Since PET between the donor and C60 can take place through space, they can be connected by means of non-covalent interactions, giving photoactive supramolecular couples. If C60 is properly functionalized, combined with the presence of central metal ions within the different heterocycles, the two units can be connected directly by metal complexation. The use of non-covalent interactions have shown to be a very useful tool to access very complicated architectures with less effort, including others that otherwise could not be approached through simple covalent chemistry.
In the following article we would like to illustrate the high versatility of axial coordination to produce structurally complex dyads from fairly simple building blocks, focusing on how structural modifications can tune the properties of such systems. The review starts with the description of the most popular systems based on pyridine coordination with metal porphyrins. Since this strategy can be applied to other heterocycles, the review continues with some recent examples of axial coordination between aromatic alkoxides and boron subphthalocyanines. The most interesting photophysical properties observed for each system are also described. Detailed reviews in this field have been recently published by D'Souza and Ito [11,12].
2 Porphyrin–pyridyl complexes
Pyridine is known to coordinate a wide variety of metal porphyrins [13,14]. The efficiency of coordination depends mainly on the metal ion. Following this approach, dyads between fullerenes and metalloporphyrins by axial coordination have been described.
C60 was functionalized via [3 + 2] cycloaddition with a pyridyl coordinating group using sarcosine and 4-pyridine aldehyde as the starting materials [15,16] yielding fullerene ligand 1. Several dyads have been assembled between ligand 1 and zinc and ruthenium tetraphenyl porphyrins (Scheme 1) [17–19]. The difference between both metalloporphyrins relies mainly on the strength of the metal–N coordination bond. In the case of zinc tetraphenylporphyrin (ZnTPP), the coordination with pyridine is weak since the only σ bonding is present. While in the case of ruthenium tetraphenylporphyrin (Ru(CO)TPP) σ bonding is reinforced by π backbonding.
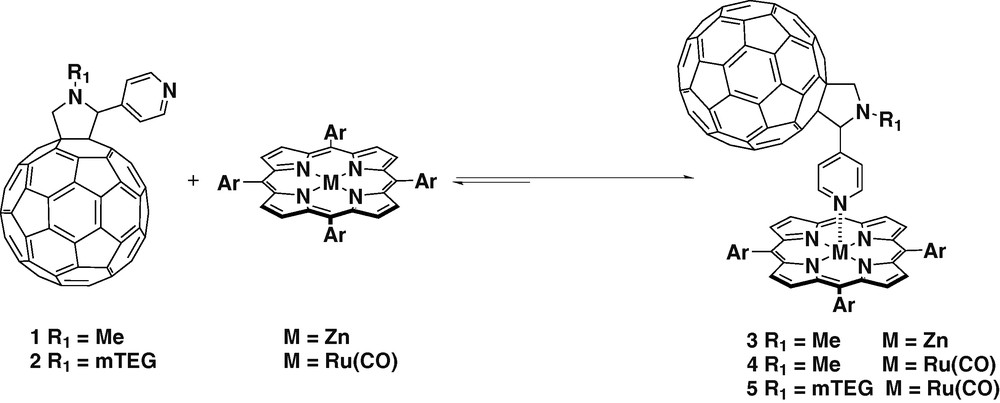
The coordination between ZnTPP and ligand 1 was extensively studied [18,19]. The absorption spectrum of ZnTPP was measured at increasing concentrations of the fullerene ligand 1 in different solvents. In non-coordinating solvents (e.g. toluene, dichloromethane) the spectra revealed a red shift of the Q bands and a broadening of the Soret band, which was attributed to the formation of the complex. In more polar and coordinating solvents (e.g., THF, benzonitrile) such effect was not observed, indicating that there was competition in the coordination of zinc between the fullerenic pyridine and the solvent. 1H-NMR experiments were carried out with 1:1 mixtures of the ligand 1 and ZnTPP in CDCl3, the pyridine protons showed shifting from 8.4 to 3.6 ppm due to anisotropy effected by porphyrin ring over the pyridine substituent confirming that the coordination with the zinc atom occurred. The photophysics of complex 3 showed intramolecular PET. In dichloromethane, a lifetime of 8.6 μs was observed. While in polar solvents such as benzonitrile very efficient charge separation was detected with lifetimes of several hundreds of microsecond. The differential absorption changes observed in benzonitrile, revealed the participation of two components in the formation of C60 radical-anion. One corresponds to intramolecular PET when the complex is assembled. The other one was assigned to the intermolecular PET between the free ligand and the porphyrin. This effect was also observed in EPR experiments.
Similar experiments were carried out with Ru(CO)TPP (Scheme 1) [19]. Complex 4 showed primarily intramolecular energy transfer (EnT) from the photoexcited porphyrin to the fullerene in toluene. In benzonitrile, in which the increase of polarity enables bond detachment, intramolecular charge-separated states followed by fast recombination were observed. The electrochemical behavior of 4 was studied by cyclic voltammetry (CV) [20]. The cathodic pattern showed eight one-electron reversible reduction waves. Based of CV experiments on ligand 1 and on Ru(CO)TPP, five reductions were assigned to fullerene centered reductions while the other three correspond to porphyrin reduction processes.
Complex 5 was prepared using ligand 2 and Ru(CO)TPP (Scheme 1) [20]. Ligand 2 is characterized by a methyltrietheleneglycol chain on the nitrogen atom of the fulleropyrrolidine. The introduction of such a hydrophilic chain allowed the preparation of Langmuir–Blodgett films. The structure of 5 did not provide the adequate hydrophobic–hydrophilic balance to ensure the formation of stable Langmuir monolayers. The floating films were prepared using arachidic acid as an additive, giving reproducibility in the preparation of the film and more importantly, the dyad molecules were separated favoring intramolecular processes. The layers were transferred to solid substrates and studied by steady-state and transient asbsorption spectroscopy. Again the radical-anion of C60 was detected with a lifetime of 2.2 μs.
Analogous systems with C60 functionalized via Bingel reaction bearing a coordinating pyridyl group have been reported [21]. The association constant (Ka) with ZnTPP was determined by 1H-NMR (Ka = 3600) and fluorescence titrations (Ka = 3000). EnT was observed by time resolved luminescence spectroscopy.
A supramolecular dyad with a quasilinear geometry involving an azafullerene and a substituted ZnTPP, was prepared and studied by Guldi and Hirsch [22]. In toluene photoinduced singlet-singlet EnT was observed while in o-dichlorobenzene PET was detected (Fig. 1).
Azafullerene–porphyrin dyad with a quasilinear geometry.
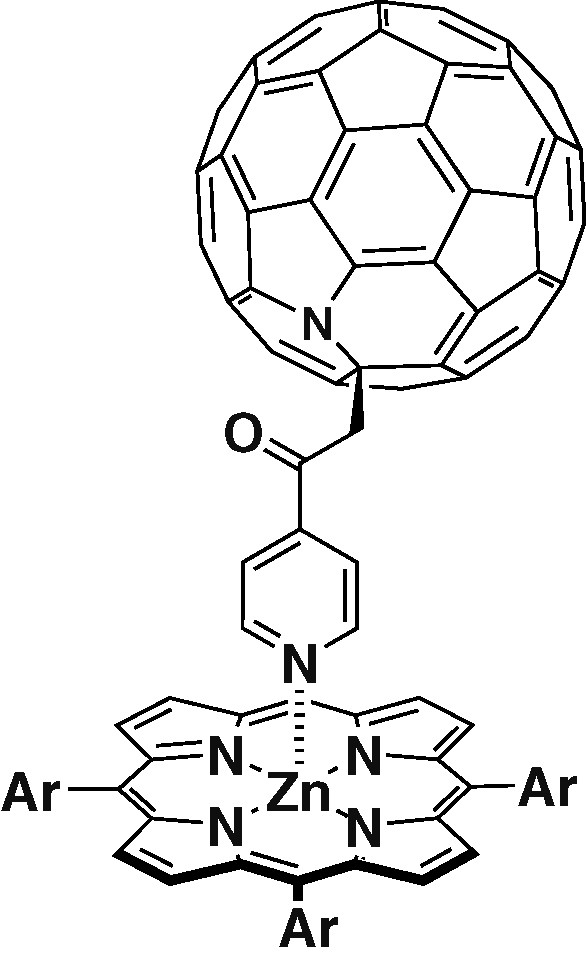
A triad formed between the ligand 1 and a parachute-like fullerene–zinc porphyrin dyad 6 is depicted in Scheme 2 [23]. Selective irradiation of the porphyrin in both toluene and benzonitrile did not show any changes when compared with the PET observed for dyad 6 by itself. Upon irradiation in the wavelengh of fullerene absorption, PET for 7 was activated. Since benzonitrile was used as a solvent, the equilibrium between the complex and non-complexed species is affected. This divided the reaction into two processes. A fast intramolecular reaction between the singlet excited state of fullerene in the coordinated complex 7 followed by a much slower intramolecular reaction between the fullerene triplet excited state and the free dyad 6 (Scheme 2).
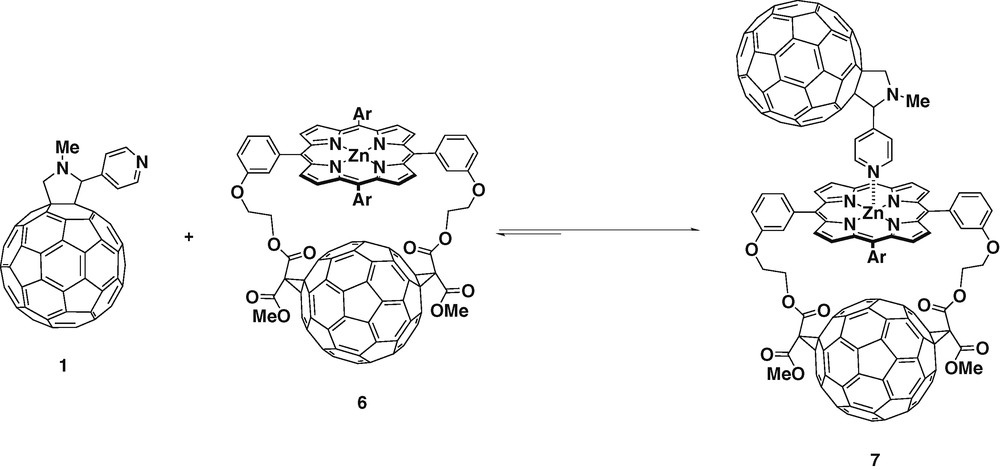
Structural variations can be easily introduced in the fullerene ligands thanks to the versatility of the [3 + 2] cycloaddition of azomethine ylids to C60. This allows the study of how structural variations may affect to the charge separated state. When using glycine and 3-pyridine aldehyde as starting materials, meta-substituted fulleropyridine ligand 8 was obtained (Scheme 3) [24].
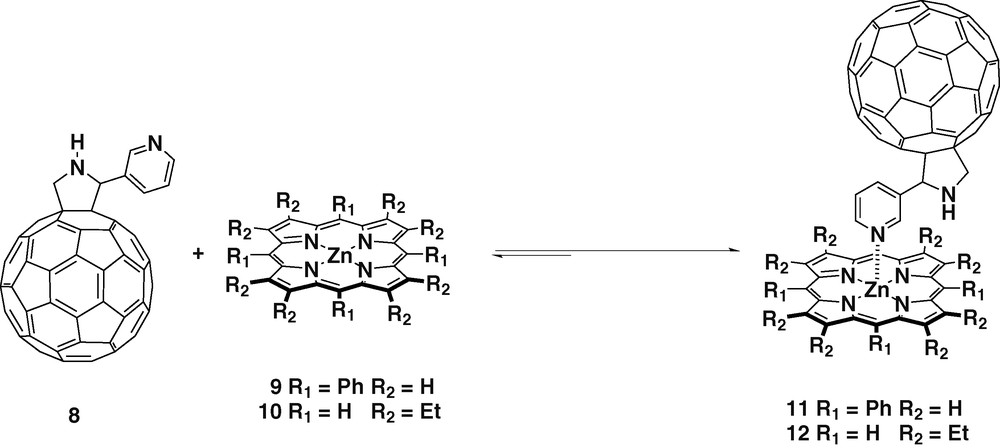
Again UV and 1H-NMR studies confirmed the formation of the complexes 11 and 12 when using zinc porphyrins 9 and 10, respectively, revealing a 1:1 stoichiometric ratio between the donor and the acceptor. In addition, single-crystal X-ray diffraction of 11 confirmed the complexation undoubtedly [25]. The equilibrium constants calculated together with the oxidation potentials of the zinc tetrapyrrole revealed stable complexations. The fluorescence of the porphyrin was 60% quenched in complexes 11 and 12, compared with the free porphyrins 9 and 10, this effect was assigned to the occurrence of efficient PET from the excited zinc porphyrin to C60.
Ligand 13 was obtained when N-pyridylglycine and paraformaldehyde were used as the precursors for the azomethine ylide. The pyridyl group was attached directly to the nitrogen of fulleropyrrolidine giving the highly symmetric ligand 13 [26] (Scheme 4).
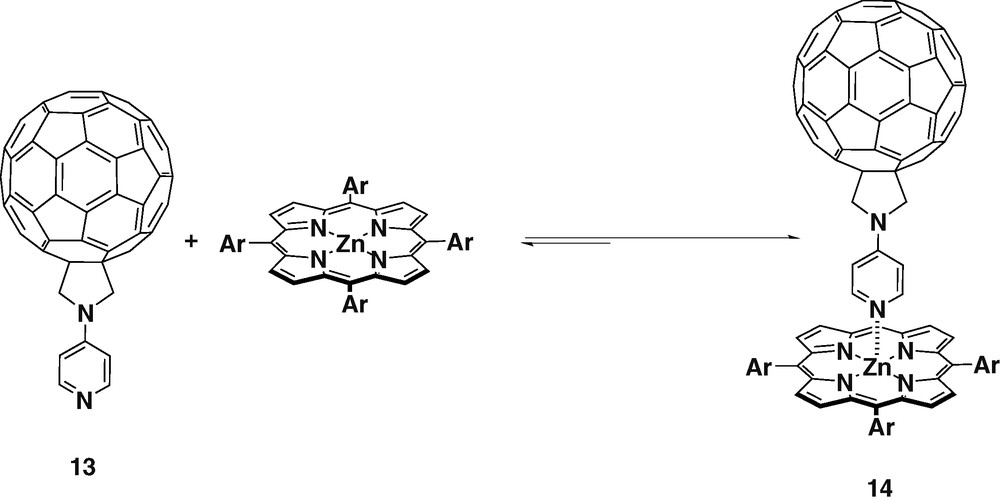
Molecular modeling performed at the semi-empirical AM1 level using SPARTAN showed that ligand 13 could form linear complexes with ZnTPP. The charge (Mulliken) was calculated for ligands 1 and 13. The results showed that the density on the nitrogen of the pyridyl group in 13 was higher, therefore it should bind more stongly with ZnTPP. The complexation of ligand 13 and ZnTPP was monitored by 1H-NMR spectroscopy has shown that in the 1:1 mixture, the pyridyl protons signal was shifted upfield by 5.94 ppm. This very large NMR shift of the pyridyl protons and the absence of the peaks of the uncomplexed ligand 13 confirmed the strong complexation between the both species.
Complex 14 was studied by time-dependent EPR experiments [27], revealing that at low temperatures in frozen matrices, photoexcitation of the ZnTPP donor results mainly in singlet–singlet EnT to the pyridine appended fullerene acceptor. On the contrary, in fluid phases, PET is the dominant process. Therefore, in isotropic solvents, the generated radical pairs were long-lived, with lifetimes exceeding those observed for covalently linked donor–acceptor systems. Meanwhile, in the case of anisotropic nematic liquid crystals no long-lived radical pairs were created. This can be explained by the fact that in ordered liquid crystal matrix, the separation of the complex constituents is hindered, and as a consequence fast intramolecular back electron transfer takes place. These experimental results confirm the existence of equilibrium between association and dissociation of the coordinatively linked complex. The electron or EnT occurs in polar as well as nonpolar solvents.
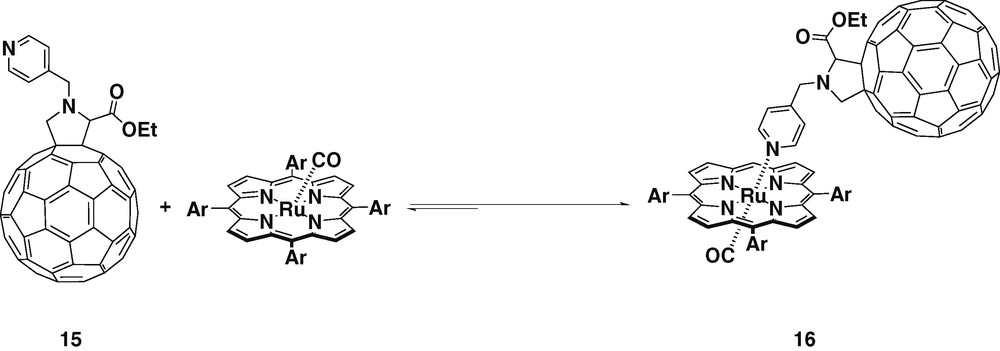
The synthesis and conformational analysis of a new ruthenium porphyrin–fullerene donor–acceptor ensemble (16) has been reported [28] (Scheme 5). Again, the pyridyl group was directly attached to the nitrogen of the fulleropyrrolidine but with an extra carbon atom between them. This new geometry can imply either a longer or a shorter donor–acceptor distance if the ligand is in an extended or in a bent conformation, respectively. Conformational studies were performed in order to verify the preferred geometry, and the bent conformation, in which the fullerene gets close to the porphyrin, was found to be the most stable. All additional analyses and calculations carried out on other conformers confirmed the tendency of the molecule to arrange itself in the bent conformation. Steady-state and time-resolved emission experiments indicated that this new donor–acceptor dyad gives rise to rapid intramolecular deactivation of the ruthenium porphyrin triplet excited state, depending on the solvent polarity. In fact, in nonpolar solvents, as toluene, a rapid triplet energy transduction occurs, while in THF and benzonitrile, PET was observed. The lifetime of this charge-separated state was found to be on a timescale of 50 μs, which is remarkable since similar spaced donor–acceptor systems are subject to intramolecular charge recombination of the order of a few nanoseconds.
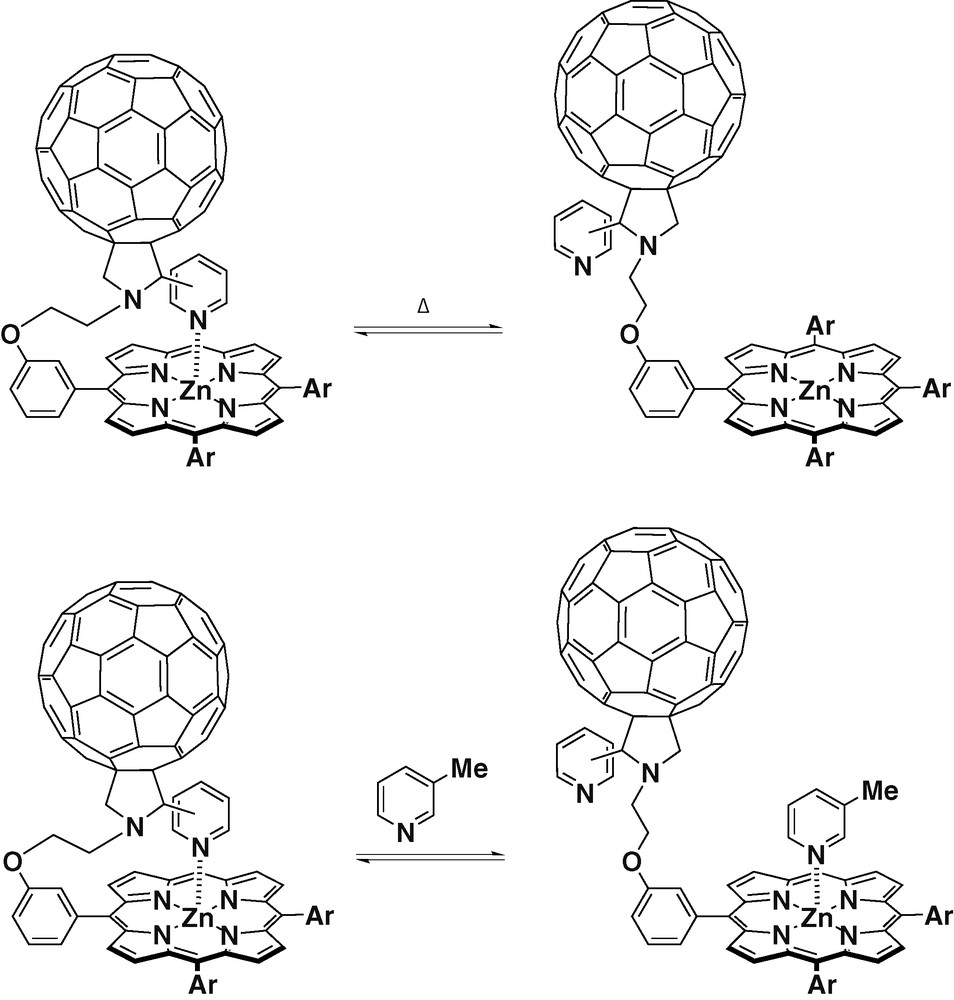
D'Souza et al. [29] have reported the synthesis and studies of dyads with meta and para substitution patterns in the pyridyl group, covalently linked to one of the phenyl rings of a ZnTPP. These dyads have shown to be capable of intramolecular axial coordination between the N-pyridyl ligand and the ZnTPP, via a ‘tail-on’ and ‘tail-off’ binding mechanism (Scheme 6). These derivatives constitute a novel approach to position the donor zinc porphyrin and the acceptor C60 in a defined spatial organization, and the donor–acceptor proximity can be controlled either by temperature variation or by an axial ligand replacement method as pictured in Scheme 6. UV–visible absorption spectroscopy experiments realized at room temperature for the ZnTPP–C60 dyads, showed that the Soret bands were located at around 433 nm compared to a 425-nm Soret band of ZnTPP alone. These results showed the existence of a pentacoordinated zinc porphyrin due to the axial coordination of the pyridyl unit. Further concentration dependence studies revealed no changes, indicating the absence of any intermolecular interactions. The effect of ‘tail-on’ and ‘tail-off’ forms has been probed by both steady-state and time-resolved fluorescence emission. The efficiency change in charge separation occurring in the ‘tail-off’ form in comparison with the results obtained for the ‘tail-on’ form, has suggested the presence of some through-space interactions between the singlet excited ZnTPP and the C60 moiety in the ‘tail-off’ form. Furthermore, in these studied dyads, a long-lived ion pair with a lifetime of about 1000 ns was observed.
In a recent example [30], fullerene was functionalized with two pyridyl coordinating groups (Fig. 2). This new ligand can bind up to two porphyrins. The binding experiments carried out showed that the ligand could coordinate more effectively the porphyrin dimer shown in Fig. 2 than two ZnTPPs. Steady-state and time-resolved fluorescence studies revealed efficient charge separation in the nanosecond scale.
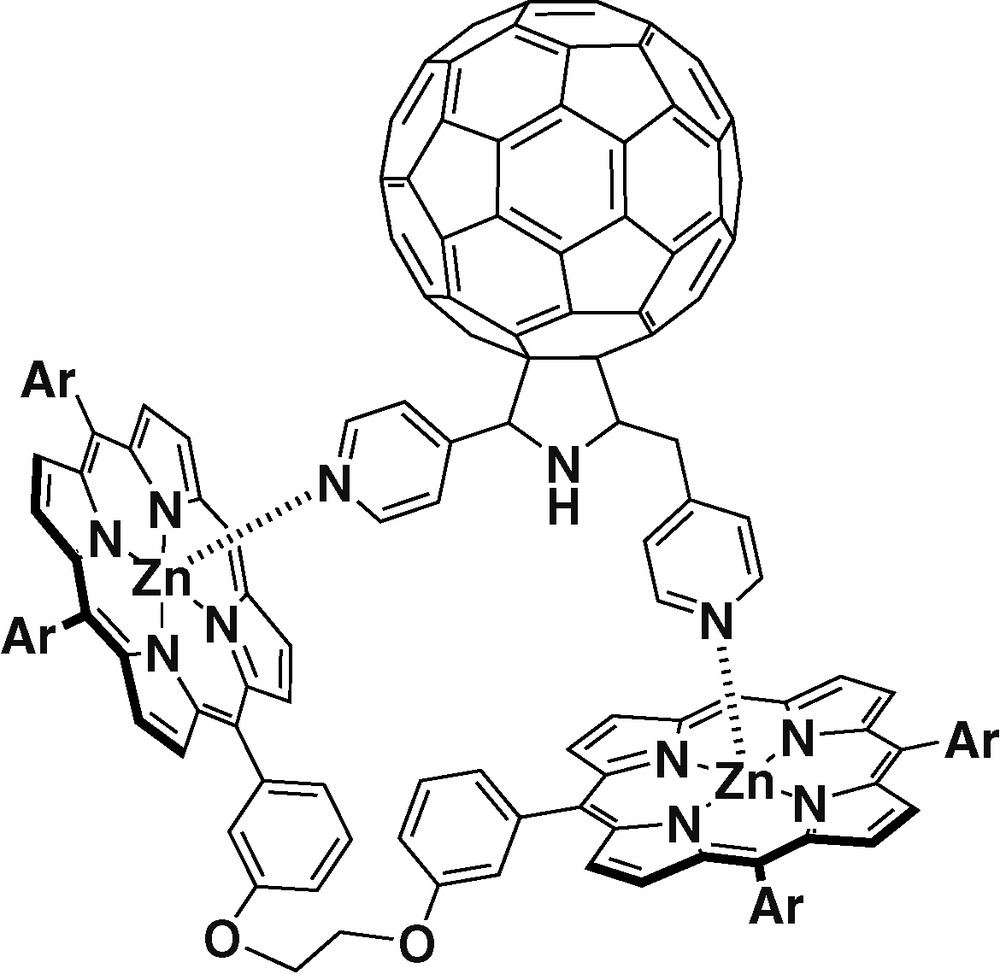
Dipyridyl fullerene ligand complexed with a porphyrin dimer.
3 Subphtalocyanine–phenoxide complexes
The subphthalocyanines (SubPcs) are nonplanar aromatic macrocycles comprising three N-fused diiminoisoindole units and an axial ligand around a tetracoordinated boron atom. They are synthetically versatile molecules and possess good characteristics for efficient intramolecular electron or energy transfer reactions.
Complexation has also been applied to prepare fullerene-subphthalocyanine couples, by introducing 2-phenoxy substituents in C60. Gonzalez-Rodriguez et al. [31] have reported the synthesis and photophysical properties of a series of three subphthalocyanine-C60 dyads in which the two subunits are placed at different distances in function of the phenyl substitution from ortho to meta to para position (Fig. 3). UV–visible spectra of the dyads revealed intramolecular interactions in the ground state between the two subunits. Electrochemistry of the dyads has indicated that the meta substituted 17b was the easiest to reduce and to oxidize, indicating also the weakest ground-state interaction between the donor and acceptor groups in the series. These results suggest that the electronic communication took place through-bond and not through-space. Fluorescence studies have demonstrated that a singlet–singlet EnT dominates over PET.
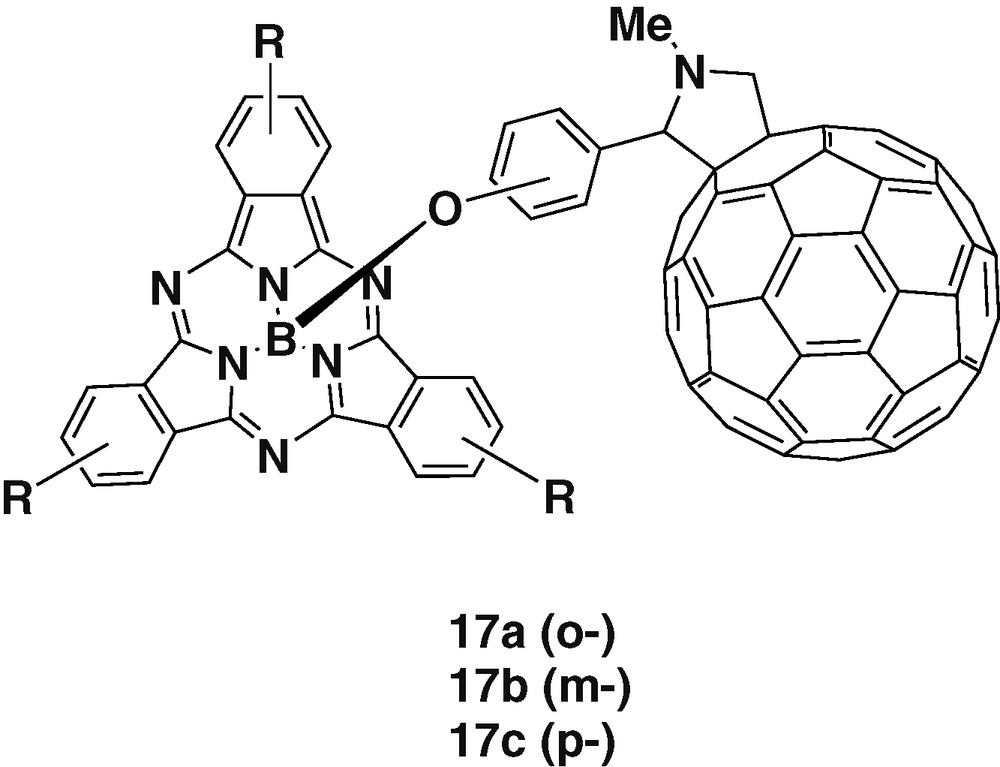
Subphthalocyanine alkoxide complexes with different fullerene ligands.
Further studies have been carried out on four subphthalocyanine–C60 dyads 18a–d, pictured in Fig. 4. These derivatives have been prepared with substituents of different electronic character, such as fluorine or iodine atoms and ether or amino groups [32], allowing a fine tuning over the electron-donating properties of the resulting dyad. Electrochemistry measurements carried out on the different compounds revealed that the nature of the peripheral substituents present on the macrocycle modifies strongly the reduction and oxidation potentials of the subphthalocyanines. The use of such different substituents provided the means to control the interplay between energy and charge transfer processes. Then the possibility of tuning the properties of the final dyad by introducing different groups at the periphery of the subphthalocyanine, proved the versatile character of this chromophore.
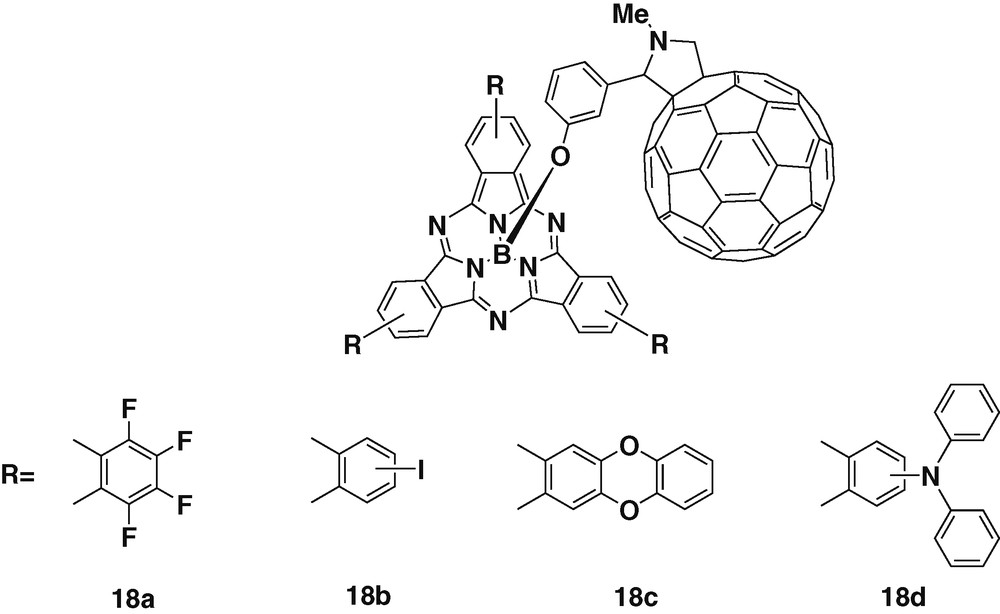
Subphthalocyanine – fullerene dyads with suphthalocyanines displaying different substituents.
4 Conclusions
Axial coordination to metals is an excellent way to construct novel supramolecular photoactive architectures. The use of such interaction to couple fullerene with a wide range of electron-donating heterocycles such as porphyrins, subphtalocyanines provides a very useful model in creating/designing artificial photosynthetic systems. Efficiency of coordination depends mainly on the metal ion. The properties can be modified by small structural variations introduced covalently on the building blocks can tune the properties of the assembled systems, and also by external factors such as temperature, solvent polarity or the nature of matrices used. All these modification parameters can interfere on the photophysical properties and then can influence the electron or energy transfer occurring between the entities. Also complexation between subphthalocyanine and fullerene has shown to give very versatile new dyads. The introduction of different kinds of electronic substituents allows fine-tuning of the electron donating processes and properties.
Acknowledgments
The authors gratefully acknowledged the European Union (RTN-EMMMA and RTN-CASSIUSCLAYS) and MIUR (PRIN 2004, prot. 2004035502) for funding and support of this work.