

1 Introduction
Soon after the discovery of a method to produce [60]fullerene in bulk quantities [1], scientists worldwide began exploring its fascinating chemistry. One of the very first reactions investigated was that between [60]fullerene and aliphatic amines [2]. Primary and secondary aliphatic amines add readily and repeatedly across [60]fullerene. Tertiary aliphatic amines were soon thereafter shown to undergo photocycloadditions across [60]fullerene [3]. Despite the potentially rich chemistry available via reaction with a wide range of aliphatic amine structures, a detailed understanding of the chemistry between aliphatic amines and [60]fullerene was slow to develop. This was due to the difficulty in characterizing the frequently complex product mixtures as well as a poor understanding of the reaction mechanisms involved. Slowly but surely, these obstacles are lifting. Recent synthetic advances include conditions for the formation of well-defined tetraamino epoxide adducts in excellent yield [4]. Mechanistic insights include the realization that singlet oxygen plays a significant role in the photocycloaddition of tertiary amines [5], and perhaps in other reactions as well. Closely related to aliphatic amine additions are the reactions between [60]fullerene and aliphatic polyamines. At low temperatures, the chemistries involving polyamine are quite similar to their mono- and diamine counterparts. At elevated temperatures, however, polyamines have been shown to be useful reagents for the preparation of highly hydrogenated [60]fullerenes [6]. Diethylene triamine in particular is an effective hydrogenation reagent for the regioselective formation of C3v C60H18 in excellent yield.
2 Addition of primary and secondary aliphatic amines
Wudl et al. were the first to report the rich chemistry between [60]fullerene and aliphatic amines like n-propylamine, n-dodecylamine, tert-butylamine, and morpholine [2]. These and other primary and secondary aliphatic amines add readily and repeatedly across [60]fullerene producing complex reaction mixtures composed of numerous structures and isomers. Each amination reaction reportedly adds the elements R(R′)N and H across a π-bond on [60]fullerene (Fig. 1). A number of regiochemistries are possible but available evidence (vide infra) suggests that hydroaminations prefer either a 1,2- or a 1,4-addition. The number of amine additions seems to be a function of the amine, the conditions of the experiment, and the solubility of the resulting products. Thus, products from the morpholine reaction precipitate from neat morpholine solution after approximately six additions [2,7] while smaller amines like n-propylamine add a dozen times without precipitation. Aqueous solutions of dimethylamine have been utilized to add approximately 10 equivalents of dimethylamine under biphasic conditions (H2O/CHCl3) without precipitation of product [8]. Unless oxygen is rigorously excluded, it will be incorporated into the product structures (vide infra).
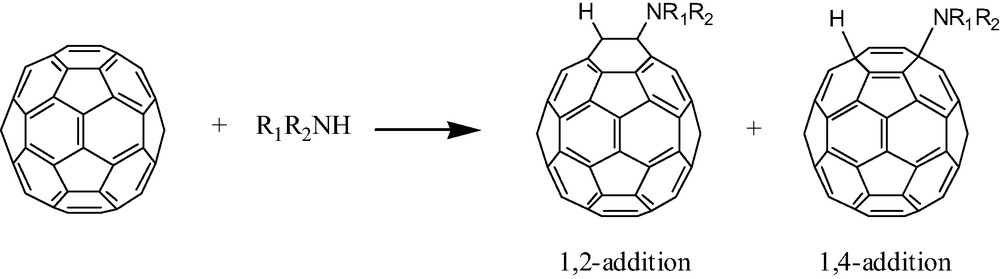
Hydroamination of [60]fullerene with primary or secondary aliphatic amines.
Soon after the discovery of this potentially rich chemistry, a morpholine adduct of [60]fullerene with stated formula C60H6(morph)6 was reported to undergo multiple, rapid, nearly degenerate [1,5]-sigmatropic hydrogen shifts (i.e. ‘globe-trotting’ hydrogen) on the [60]fullerene surface [7] as judged by variable temperature NMR studies. This dynamic behavior, however, was later shown to be due to temperature and dilution dependent hydrogen bonding interactions between amine moieties on the fullerene surface and residual water, not globe-trotting hydrogen. Thus, Miller et al. demonstrated [8] that the NMR signals attributed to globe-trotting hydrogens could be completely removed upon the addition of 3 Å molecular sieves and returned again upon spiking samples with additional water. Moreover, dilution dependencies ruled out an intramolecular process.
While the complex product mixtures generated upon reacting [60]fullerene with excess primary or secondary aliphatic amine are problematic to characterize, Kampe et al. reported a simple method [9] for the generation of well-defined amine addition products using secondary diamines like N,N′-dimethylethylenediamine or piperazine (Fig. 2). The chemistry utilizes a modest excess of diamine and produces both mono- and bisadducts in overall 50–85% corrected yields (i.e. corrected for total quantity of [60]fullerene consumed). The bisadducts thus formed have been carefully isolated and characterized [9,10]. Butts and Jazdzyk later demonstrated the photocycloaddition of substituted piperazines in good yield [11]. They demonstrated that substituted piperazines can be employed for the introduction of a large variety of additional functional groups including alcohol, ether, amide, and ester groups. Interestingly, the products of diamine addition, whether prepared by thermal or photochemical means, are not hydroamination products as they possess no fullerenic C–H bonds. Are fullerenic C–H bonds initially formed in these reactions only to be oxidatively removed in a subsequent step? There is no direct evidence for this pathway but it seems likely given that Leigh and co-workers demonstrated formation of three well-defined hydroamination monoadducts possessing new fullerenic C–H and C–N bonds [12]. Bulky aza-crown ethers (secondary amines) were employed resulting in both 1,2- and 1,4-hydroamination products. While the 1,4-addition pattern introduces an unfavorable 6,5 double bond into the structure (see Fig. 1), this product may nonetheless be energetically competitive given the bulky nature of the aza-crown ethers and the somewhat more sterically demanding nature of the 1,2-addition pattern. 1H-NMR resonances for the fullerenic C–H hydrogens were observed at approximately 5.8 ppm, similar to the chemical shifts for other fullerenic C–H hydrogens on related molecules [13]. If fullerenic C–H bonds do indeed form in the diamine reactions, their rapid oxidation in air would not be surprising. Oxygen appears to play a significant role in several aliphatic amine reactions (vide infra).
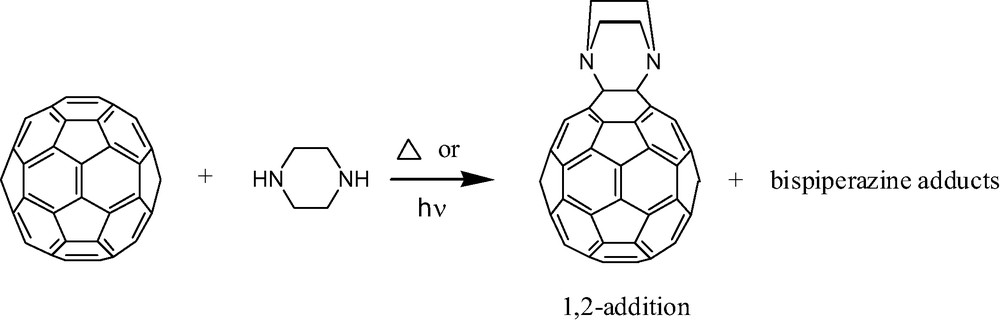
A 1,2-addition of piperazine across [60]fullerene. See Refs. [9–11].
Unlike the case with secondary amines, no well-defined adducts of primary amines (excluding polyamines) have been reported thus far. This has not prevented, however, the exploitation of primary aliphatic amine chemistry for the preparation of interesting materials including fullerene grafted polymers [14] (Fig. 3).
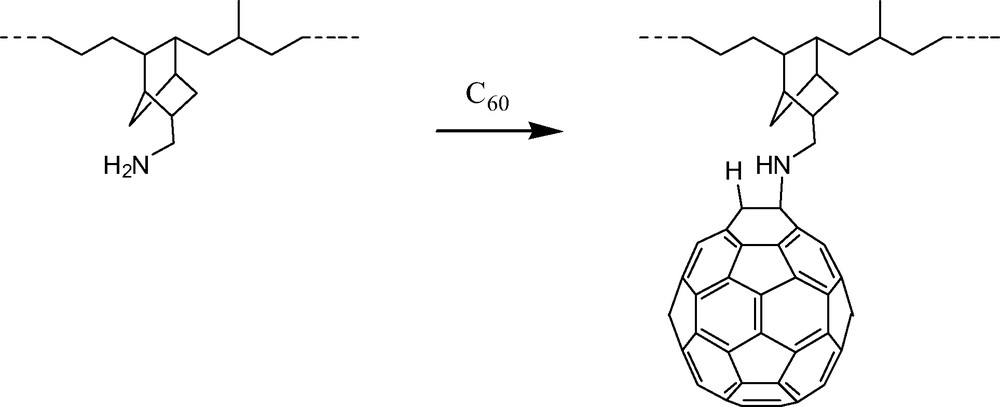
Preparation of a [60]fullerene grafted polymer using amine functionalized ethylene propylene terpolymer (EPDM–amine). See Refs. [14a, b].
3 Mechanism of primary and secondary aliphatic amine addition to [60]fullerene
Considerable evidence exists that radical ion-pairs form when [60]fullerene is mixed with aliphatic amine. Wudl et al. first noted the formation of an EPR active solution upon adding [60]fullerene to aliphatic amine [2]. Skiebe et al. [15] and others [16] followed this observation with careful spectroscopic studies (EPR and UV–vis–NIR) of the radical ion-pairs formed. Does the mere existence of fullerene radical anion–amine radical cation pairs necessarily implicate these species in the amination mechanism? The answer must be ‘no’. But while the formation of radical ion pairs could in principle be independent of the amination mechanism, several observations seem to link the two tightly together: (a) [60]fullerene reacts rapidly with many neat solutions of primary or secondary aliphatic amine in the dark; (b) [60]fullerene reacts only sluggishly with the same primary or secondary aliphatic amines when reactions are performed in relatively nonpolar solvents like toluene or chloroform in the dark; (c) [60]fullerene reacts rapidly again with many of the same primary or secondary aliphatic amines when reactions are performed in relatively nonpolar solvents like toluene or chloroform in the presence of light. These observations are most easily interpreted to indicate that the first step in fullerene amination involves a solvent dependent electron transfer from amine to [60]fullerene. Because electron transfer processes of this type are slower in non-polar solvents, photoexcited [60]fullerene—a better electron acceptor—facilitates reactions in nonpolar solvents.
Initial electron transfer from amine to [60]fullerene should be followed by either radical coupling to produce a zwitterionic intermediate (path a, Scheme 1) or a proton transfer within the contact radical ion pair to produce a pair of neutral free radicals (path b, Scheme 1). Support for this latter path is garnered from the photochemistry literature where primary and secondary aliphatic amines are known to react with photoexcited trans-stilbene to produce hydroamination products. These reactions proceed via proton transfer in the contact radical ion pairs followed by radical combinations [17]. Despite the higher dissociation energies associated with the rupture of N–H bonds compared to α C–H bonds, the N–H bond in the contact radical ion pair has a higher kinetic acidity. Thus, a hydroamination mechanism as illustrated in path b of Scheme 1 seems reasonable. Nonetheless, other mechanisms cannot be ruled out without further study. In particular, hydroamination could proceed via a singlet oxygen path as proposed for the photocycloaddition of tertiary amines [5] (vide infra). A direct nucleophilic addition to produce the same zwitterionic intermediate shown in Scheme 1 also seems plausible but is not consistent with the light requirement noted above. Additional work is needed to further test these mechanistic possibilities.
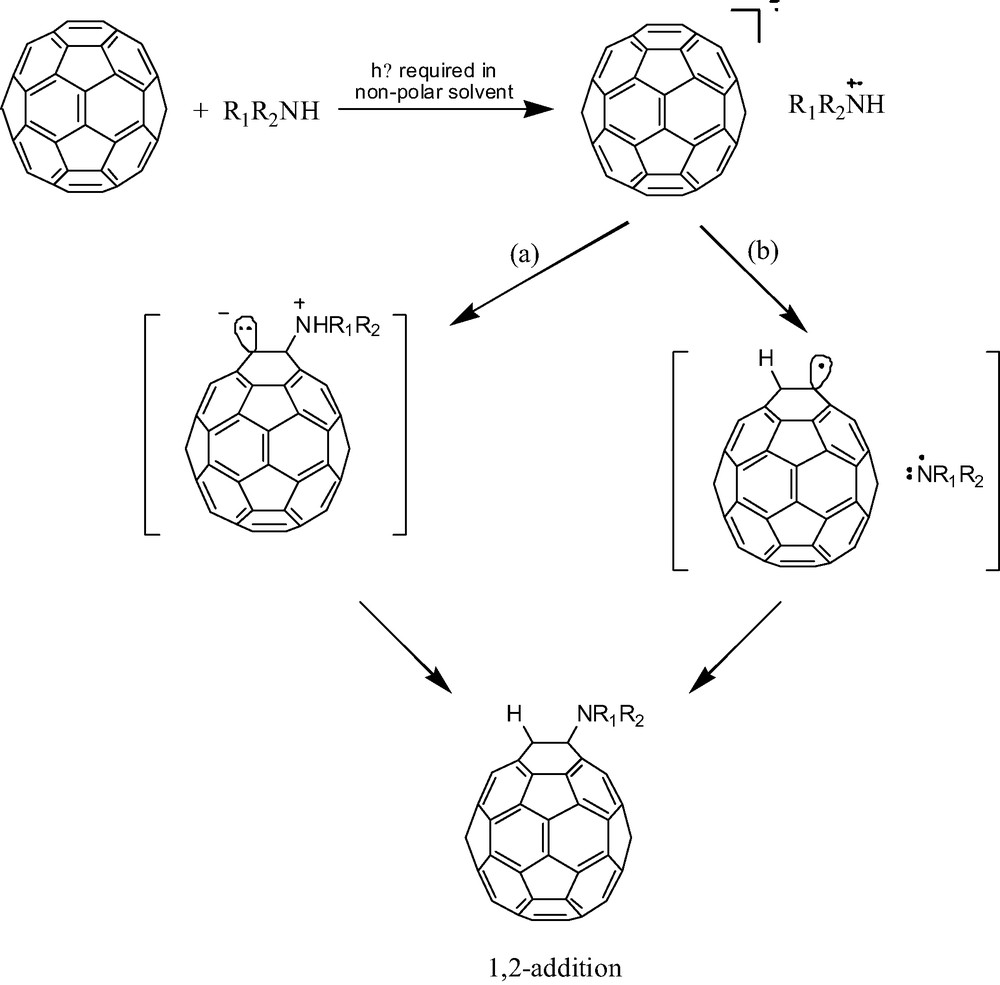
Proposed mechanism for hydroamination of [60]fullerene in the absence of oxygen.
4 Photocycloadditions of tertiary amines
Lawson et al. were the first to note [3] the photocycloaddition of a tertiary aliphatic amine, triethylamine, across [60]fullerene to produce a symmetric pyrrolidine adduct [18] (Fig. 4). Takeda et al. likely prepared the same compound [19] but did not characterize the structure. The reaction can be accompanied by the formation of unusual by-products [20] that could reduce its overall synthetic utility. Remarkably, however, the synthetic scope of this reaction has hardly been tested. Where it has been tested, results have been favorable. Guo et al. successfully applied this chemistry to the synthesis of intriguing [60]fullerene–alkaloid adducts which would be otherwise extremely difficult to prepare [21]. Bernstein and Foote prepared an alkynyl substituted pyrrolidine adduct [5] using the same approach. Interestingly, Bernstein and Foote also offered compelling evidence for a photocycloaddition mechanism involving singlet oxygen. The reaction typically requires both light and oxygen to proceed. However, when the singlet oxygen precursor 1,4-dimethylnaphthalene endoperoxide (DMNO2) was added to a reaction mixture that was kept in the dark, the same pyrrolidine adduct formed. DMNO2 thermally degrades in the dark to produce singlet oxygen without the possibility of competing photochemical pathways. The evidence is compelling and suggests a mechanism involving initial formation of singlet oxygen, 1O2, via reaction between ground state oxygen, 3O2, and excited state triplet [60]fullerene, 3C60* (Scheme 2). Fullerenes are known to be excellent sensitizers for the formation of singlet oxygen [22]. Singlet oxygen can abstract an electron from the tertiary amine to produce a radical ion-pair which in this case has no N–H proton to dissociate. Instead, α C–H deprotonation (presumably involving a second equivalent of amine as base) produces an amine radical which can add to [60]fullerene. Repeating the same sequence of steps produces the observed pyrrolidine adduct.
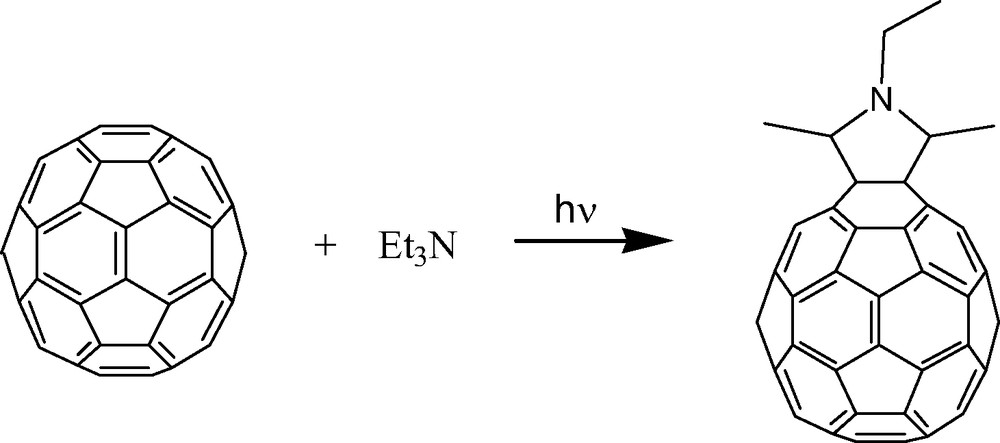
Photocycloaddition of triethylamine on [60]fullerene. See Ref. [3].
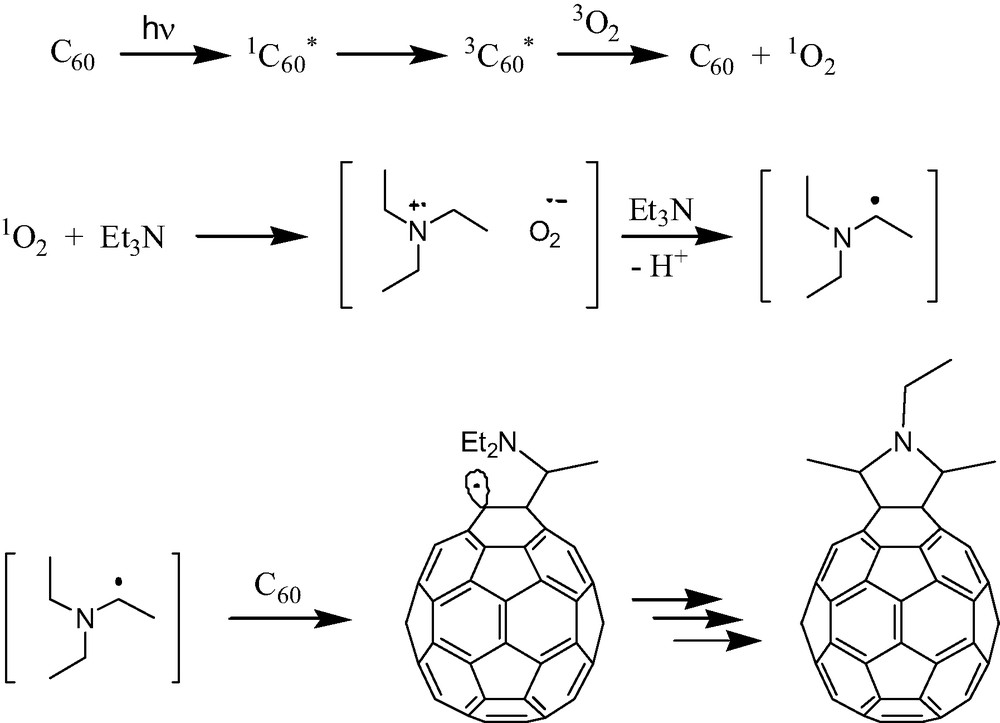
Proposed mechanism for photocycloaddition of triethylamine across [60]fullerene. See Ref. [5].
5 Regioselective aminations with secondary aliphatic amines
Early predictions that amines should add to [60]fullerene in a more or less octahedral fashion driven by the presence of six independent, reactive pyracylene units [2,7] on the [60]fullerene surface have not been borne out. Little if any data exists that may shed light on the regioselectivity of primary aliphatic amine addition to [60]fullerene. The reactions produce complex product mixtures which have thus far eluded thorough characterization. Secondary aliphatic amines, on the other hand, appear to prefer a clustered 1,4-addition pattern, particularly in the presence of oxygen. Schick et al. [23] were the first to note that the reaction between excess secondary amine and [60]fullerene yields several well-defined adducts when the reaction is run in the presence of oxygen. In this manner, bismorpholino[60]fullerene, tetrakismorpholino[60]fullerene epoxide, and morpholino[60]fullerene dimer products (Fig. 5) were all isolated and characterized. The formation of a fullerene epoxide product was not a complete surprise as oxygen atom incorporation in [60]fullerene-amine product mixtures had been previously observed by Wilson and Wu using electrospray mass spectrometry [24]. The 1,4-addition pattern, on the other hand, was not expected and this regiochemical result strongly suggests a free radical mechanism. Pioneering studies of free radical additions to [60]fullerene by Krusic et al. firmly established the existence and rationale for successive 1,4-radical additions about the periphery of corannulene substructures on the [60]fullerene surface [25]. Hirsch discusses an alternative mechanism [23] in which successive secondary amine additions are followed by 1O2 oxidation of the resulting anions (R2NC60–) or zwitterions (R2HN+C60–). However, given the regioselective addition of amines, the reaction conditions employed (i.e. large excess of morpholine) and the mechanistic insights provided by Foote and Bernstein for the photocycloaddition of tertiary aliphatic amines (Scheme 2 above), a free-radical path like that illustrated in Scheme 3 deserves careful consideration.
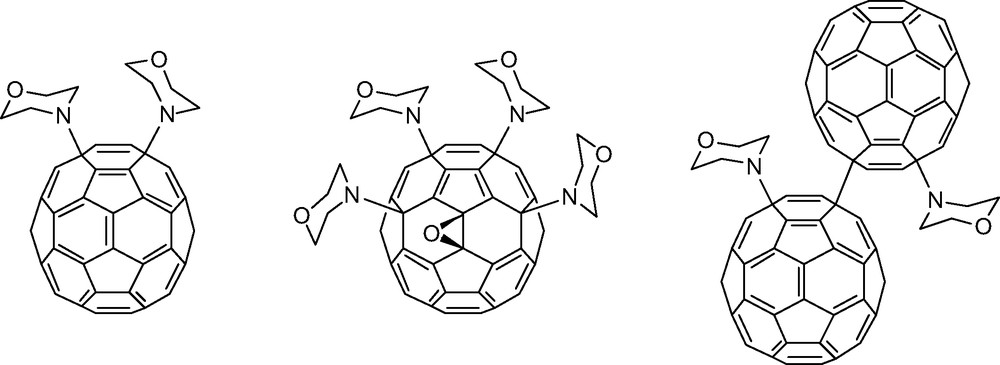
Remarkable bismorpholino[60]fullerene, tetrakismorpholino[60]fullerene epoxide, and morpholino[60]fullerene dimer products isolated and characterized by Schick et al. See Ref. [23].
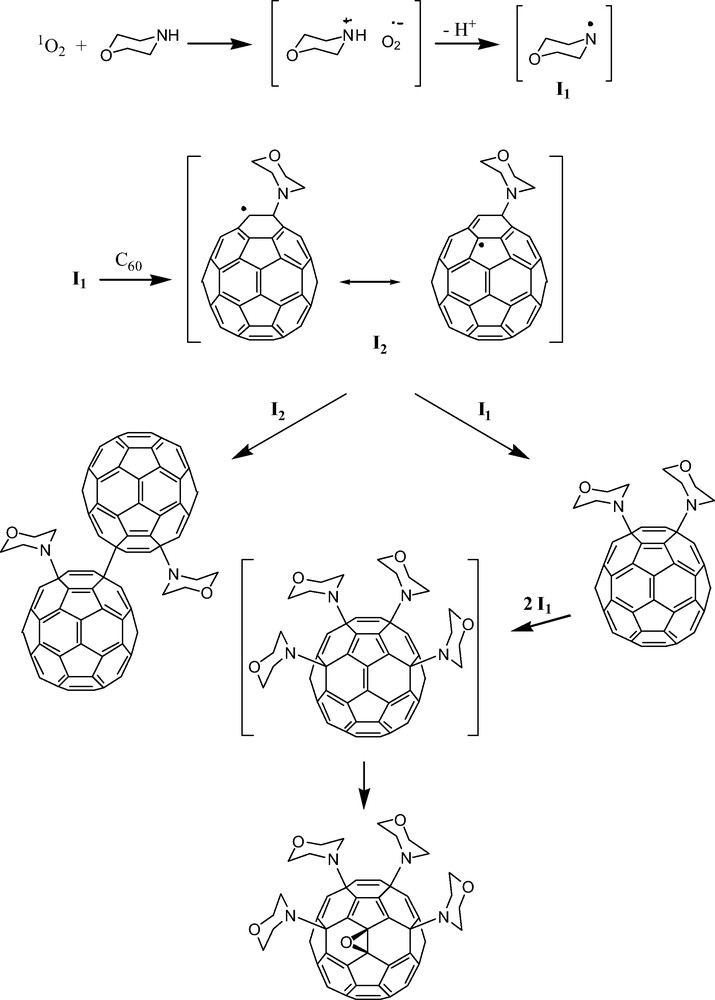
Proposed mechanism for the formation of morpholino[60]fullerene dimer, bismorpholino[60]fullerene, and tetrakismorpholino[60]fullerene epoxide.
Isobe et al. have since optimized the reaction conditions leading to tetrakisamino[60]fullerene epoxide [4,26]. In the process, they have demonstrated yields up to 98% and examined the scope of the reaction. The reaction is quite sensitive to structural effects with cyclic secondary amines giving better yields than acyclic secondary amines. Primary aliphatic amines do not give the desired product. Isobe et al. suggest a rather complex mechanism [26] involving addition of amine radical cation to neutral [60]fullerene, the formation of [60]fullerene-based carbocation, and many other steps. Application of Occam's Razor suggests that a simpler mechanism, perhaps one analogous to that illustrated in Scheme 3, should be considered. Given that Nakamura and co-workers also observe product formation when reactions are run in total darkness, another reactive oxygen species (ROS) other than 1O2 may be involved. More mechanistic work is needed for this highly interesting, synthetically useful reaction.
6 [60]Fullerene hydrogenation in aliphatic polyamines
While the chemistry between [60]fullerene and aliphatic amines has been studied steadily over the past 15 years, there has been less attention paid to the reactions with polyamines [27], save the diamine chemistry [9–11] discussed above and a highly interesting reaction observed by Isobe et al. upon mixing [60]fullerene and excess N,N′-dimethyl-1,3-diaminopropane in visible light and air saturated chlorobenzene [28]. A remarkable bis[60]fullerene adduct forms (Fig. 6) in which the N,N′-dimethyl-1,3-diaminopropane addition pattern is identical to that discussed above. A singlet oxygen path involving addition of amine radicals to [60]fullerene, analogous to that shown in Scheme 3, is implicated. Indeed, the bis[60]fullerene product is not formed in the absence of either light or air [28].
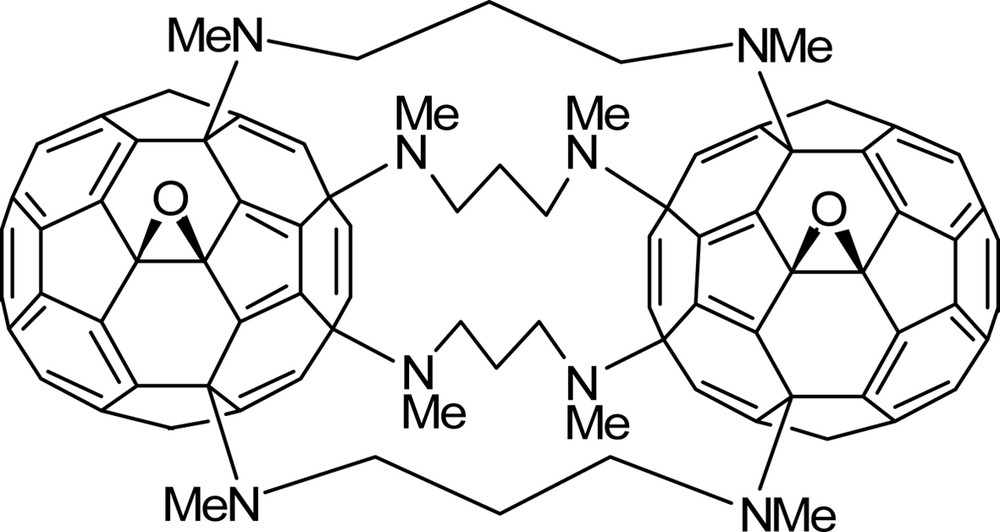
A remarkable bis[60]fullerene adduct produced by Isobe et al. upon mixing an excess of N,N′-dimethyl-1,3-diaminopropane with [60]fullerene in light and air saturated chlorobenzene. See Ref. [28].
Unsubstituted triamines and tetramines aminate [60]fullerene at room temperature to produce complex mixtures of products, analogous to reactions run in neat aliphatic monoamine. However, upon moving to higher temperatures and ultimately subjecting [60]fullerene to neat high-boiling polyamine, hydrogenated [60]fullerenes are produced in excellent yield. Briggs et al. have demonstrated [6] that reactions run in boiling diethylenetriamine produce an orange solid that analyzes for C3v C60H18 (Fig. 7). The reaction is highly regioselective and represents a simple, efficient method to produce C3v C60H18 in near quantitative yield. Moreover, the reaction is scalable to multi-gram levels. Mechanistic studies are needed to better understand this novel hydrogenation reaction.
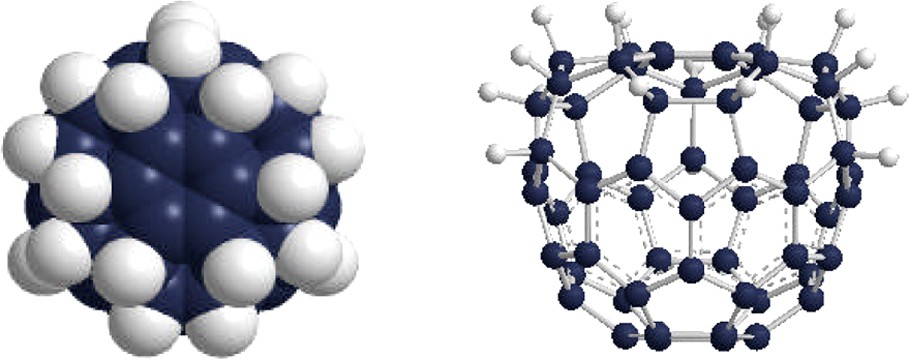
Two views of C3v C60H18 formed in near quantitative yield upon boiling [60]fullerene in diethylenetriamine. The reaction is scalable to multi-gram quantities. See Ref. [6].
7 Conclusions
A detailed understanding of the chemistry between [60]fullerene and aliphatic amines was slow to develop due to the complexity of product mixtures produced and uncertainties concerning the mechanisms of reaction. Steady progress has nonetheless been made on both synthetic and mechanistic fronts. As a result, the fullerene chemist can now prepare a variety of well-defined aminated fullerene structures in good to excellent yield. These include diamino adducts, tetraamino oxide adducts, and pyrrolidine adducts. Moreover, the chemistry can be utilized to produce interesting materials like [60]fullerene-grafted polymers, and even highly hydrogenated [60]fullerene, C60H18.
Acknowledgements
The authors thank the National Science Foundation (NSEC 0425826) and The Defense Microelectronics Activity (DMEA) for partial support of the [60]fullerene-amine research performed in our laboratories.