

1 Introduction
Despite the long standing fascination for non-planar π-extended molecules, many questions concerning these systems remain still open: can they have an aromatic behavior, how do they interact with other π-systems, what is the influence of the curvature on their electronic characteristics, do their concave and convex faces give rise to different properties?
C60 is one of these non-planar systems that behaves as both an aromatic and a non-aromatic system depending on the way it is studied. This question has generated a huge amount of work, and only recently, it was demonstrated theoretically that C60 does show weak inner and outer spherical currents that justify its ambiguous behavior [1].
On the other hand, subphthalocyanine [2] (SubPcs, Fig. 1) represents a peculiar non-planar aromatic heteromacrocycle composed of three diminioisoindole units N-fused around a boron atom presenting a 14-π-electron system. The singular structure of subphthalocyanines confers them very interesting electronic and optical properties that may be useful in the construction of artificial photosynthetic systems: (i) they are excellent antenna units that absorb in the 550–650-nm region with extinction coefficients on the order of ~50 000 M−1 cm−1 and excitation energies above 2.0 V, (ii) their rigid non-planar core affords small Stokes shifts and low reorganization energies [3], and (iii) their redox characteristics may be tuned by the introduction of different peripheral substituents.

Ball and stick representation of boronchlorosubphthalocyanine.
Due to their handy chemistry, the assembly of SubPcs into multicomponent photo- and electroactive systems may be performed via different routes involving peripheral [4] or axial functionalization as illustrated in Fig. 1 [5]. This versatility allowed the study of the interactions between photoactive and/or electroactive moieties such as C60 with both concave and convex faces of the SubPc macrocycle.
2 Convex–convex interactions in subphthalocyanine–C60 covalent assemblies
The photophysical properties of a series of SubPc–C60 dyads, in which the fullerene fragment was attached via the axial position of the macrocycle and thus, interacts through the convex face of the SubPc, have been recently studied by us [6]. Two main modifications were performed in these systems (Fig. 2): (i) the distance between the two components was modified through isomeric ortho-, meta- or para-substituted phenoxy spacers, and (ii) the macrocyclic unit was equipped with different peripheral groups (i.e. fluorine or iodine atoms and ether or amino groups), while the intercomponent distance was kept unchanged.
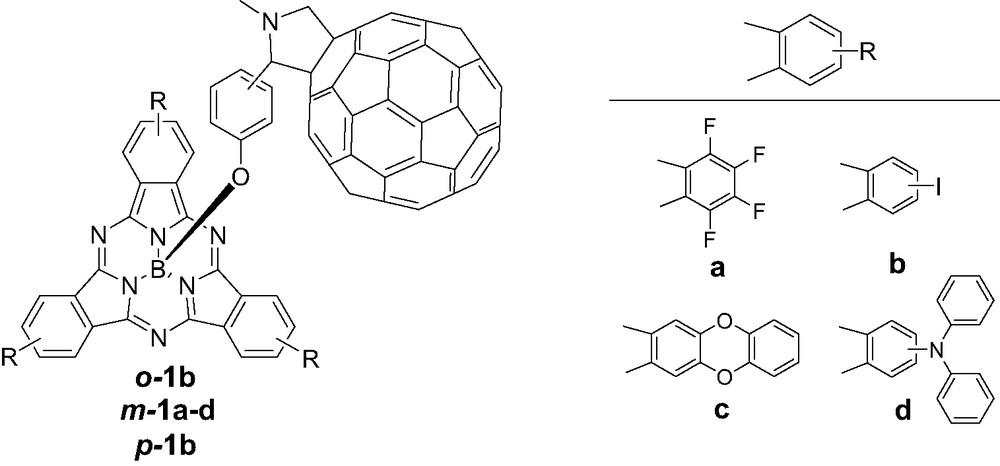
Structure of SubPc–C60 dyads o-1b, m-1a–d and p-1b.
The synthesis of the SubPc units 3a–c was carried out by the usual condensation reaction of the corresponding phthalonitriles 2a–c in the presence of boron trichloride and subsequent substitution of the axial chlorine atom with o-, m- or p-hydroxybenzaldehyde led to o-4b, m-4a–c and p-4b (Scheme 1). The preparation of SubPcs bearing electron-donating groups m-4c and m-4d initially represented a synthetic challenge, since boron halides show a strong tendency to react with amines or ether groups. We found, however, that the diphenylether moiety is stable enough to boron trichloride in the reaction conditions employed and SubPc m-4c could be obtained in moderate yields (39%). On the other hand, the introduction of amine groups was successfully accomplished by a palladium-catalyzed cross-coupling methodology between diphenylamine and triiodoSubPcs, leading to compound m-4d in good yields. In a last step, the SubPc–C60 dyads o-1b, m-1a–d and p-1b were prepared by a 1,3 dipolar cycloaddition reaction between formyl-substituted SubPcs o-4b, m-4a–d and p-4b and C60 in the conditions described by Prato and Maggini [7]. Monoaddition products were isolated in moderate yields (~45%) and, in all cases, a small amount of bisadducts (~8%) was also obtained.
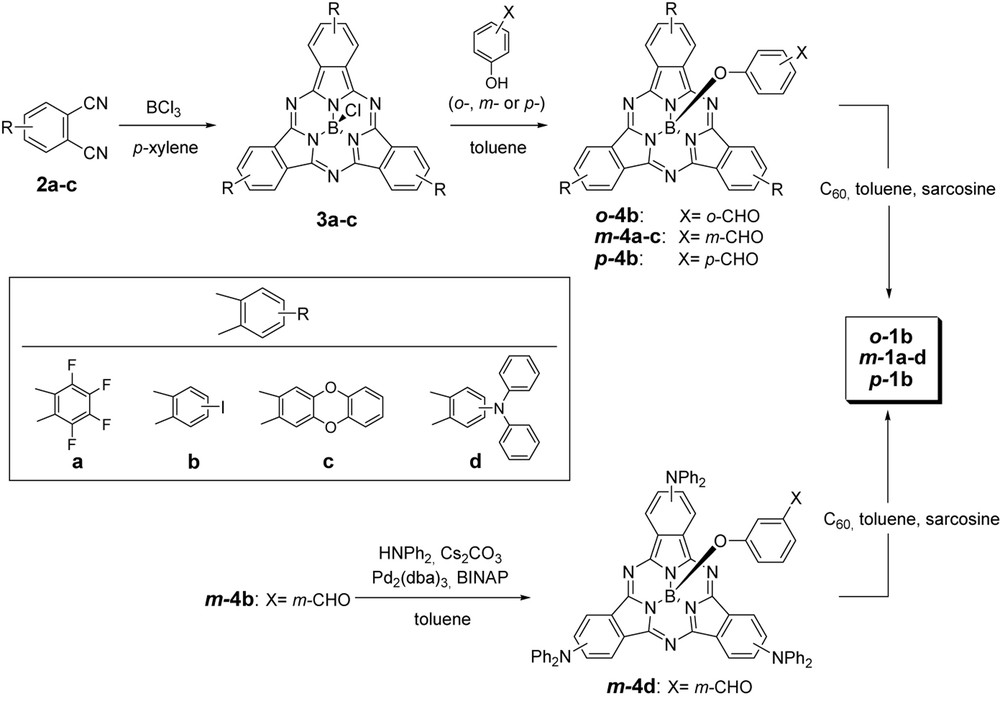
Synthesis of SubPc–C60 dyads o-1b, m-1a–d and p-1b.
The intrinsic flexibility of the phenoxy spacer, having two torsional angles that lead to multiple conformations, makes that, in solution, the C60 and subphthalocyanine units in the dyads are not restrained at fixed relative positions. The structural modeling of the o-, m- and p-systems, using molecular mechanics and AM1 semiempirical methods, allowed the evaluation of the maximum and minimum distance between the two moieties in each isomeric dyad (Fig. 3).
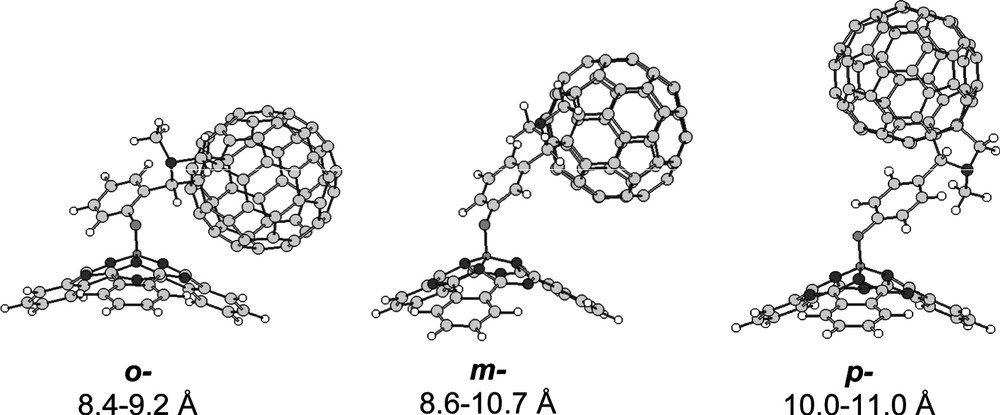
AM1 models and intercomponent distance range for o-, m- and p-isomeric SubPc–C60 dyads.
A first inspection of the absorption spectra of these systems already revealed small, but notable intercomponent ground-state interactions. In fact, the Q band of the SubPc shows a small red-shift and a broadening in the dyads when compared to the reference compounds. When comparing o-1b, m-1b and p-1b, the magnitude of this red-shift was found to be dependent on the distance between the two components, reaching a maximum for compound o-1b. Cyclic voltammetry experiments showed, however, that m-1b is the easiest to reduce and also the easiest to oxidize in this series, thus indicating the weakest ground state interaction between the donor and acceptor groups. Therefore, in the ground state, the electronic effects that dominate the different redox processes occur through-bond and not through-space, since they follow the expected (and similar) behavior for the ortho and para, and the weakest electronic coupling for the meta isomer. On the other hand, the first oxidation process of the macrocycle gradually shifts to lower potentials (by ca. 200 mV) in the series a > b > c > d, as the electron-donor character of the substituents on the SubPc increased.
The detailed photophysical characterization of these systems provided the comprehension of the different mechanistic scenarios occurring in the dyads upon initial light irradiation. Selective photoexcitation of the SubPc unit in dyads m-1a, o-1b, m-1b or p-1b, or in dyad m-1c in a non-polar environment, gave rise to a sequence of exergonic photophysical events that involve: (i) nearly quantitative singlet–singlet energy transfer to the C60 moiety, (ii) fullerene-centered intersystem crossing, and (iii) triplet–triplet energy transfer back to the SubPc. The efficiencies of these energy transfer processes were found to increase as the distance between the two components decreased. On the contrary, the stabilization of the SubPc●+–C60●– radical pair state in more polar media or by lowering the redox gap between the donor and acceptor units caused charge transfer to dominate. In the case of m-1c in benzonitrile, the thus formed radical pair decayed in 0.65 ns to yield the SubPc triplet excited state. Further stabilization was achieved for dyad m-1d, whose charge-transfer state lies now below both triplets. As a result, the charge-transfer state lifetime increased in more than two orders of magnitude (~100 ns) with respect to m-1c and showed a significant stabilization in less polar solvents, a clear attribute of the inverted region of the Marcus parabola.
3 Convex–convex interactions in subphthalocyanine dimer–C60 covalent assemblies
SubPcs are not only prone to axial functionalization, the peripheral extension of their curved aromatic surface is another possibility that has been investigated as well, which leads to SubPc analogues with substantially red-shifted absorption wavelengths. Compounds of this type include subnaphthalocyanines [8], and, more recently described, subphthalocyanine fused dimers (hereafter referred simply as Dimers) and trimers [9]. SubPc Dimers are prepared by condensation of a phthalonitrile along with tetracyanobenzene. Due to the SubPc cone-shaped geometry, the Dimers exist as two topoisomers, anti and syn, which have been isolated and unambiguously characterized. In contrast with the analogous phthalocyanine fused dimers [10], the SubPc Dimers present a distinct and highly-red shifted (ca. 120 nm) Q-band with respect to its “monomeric” counterpart.
The SubPc Dimers, while bearing two axial positions, allow a double connection to a C60-fullerene unit (in the case of the syn isomer). This double connection from the convex face of the Dimer permits an “anchoring” of the C60 suitable for through-space interactions between the acceptor C60 and donor SubPc Dimer units.
This Dimer–C60 dyad 5 (Fig. 4) was prepared [11] employing the same two-step above-mentioned procedure (axial functionalization with an aldehyde group, followed by cycloaddition to the C60-fullerene) used for the preparation of SubPc–C60 axially-linked dyads. This procedure applied to the syn topoisomer 7 yields the expected doubly-connected Dimer–C60 dyad 5 in 14% yield. The 1H NMR spectrum of the dyad 5 shows several broad signals corresponding to the multiple regioisomers and diastereoisomers (simple molecular modeling indicates that at least 20 could be formed) and the dynamic processes occurring within them. Photophysical studies reveal that the Dimer–C60 dyad undergoes a series of energy transfer events similar to those observed in the SubPc–C60 analogues m-1a, o-1b, m-1b or p-1b—after photoexcitation of the donor unit (Dimer), follows a singlet–singlet energy transfer to the acceptor fullerene, which decays by inter system crossing to the C60 triplet state, ending with the energy back-transfer to the Dimer triplet state. In contrast with the SubPc–C60 dyad, the singlet–singlet transfer in the Dimer–C60 dyad occurs between nearly isoenergetic states, as the Dimer S1 state lies at a significantly lower energy (1.77 eV) than that of SubPc (2.1 eV), thus being very close to the S1 state of C60-fullerene (1.76 eV).

Synthesis of subphthalocyanine dimer–C60 dyad.
4 Concave–convex interactions in a subphthalocyanine–C60 non-covalent assembly
Finally, we though it would be very interesting to investigate the interactions between the concave face of a SubPc macrocycle and the complementary convex surface of a C60 molecule. In order to do that, we designed a self-assembled C60 receptor 8 (Fig. 5) composed of two subphthalocyanine units connected by three pyridine–Pd(II)–pyridine linkages so that their concave surfaces would face each others and form a hollow capsule [12].
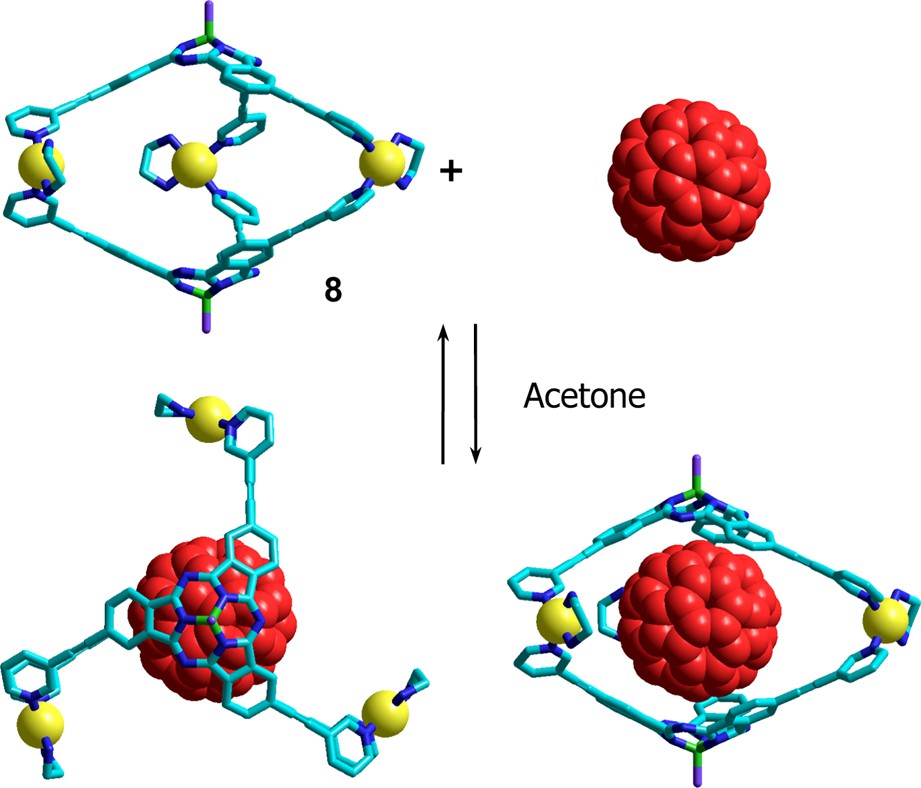
Complexation of a C60 molecule within the self-assembled subphthalocyanine cage 8.
Reaction of SubPc 3b (Scheme 2) with 3,5-ditertbutylphenol in toluene at reflux yielded the axially functionalized SubPc 9 in 80% yield. Triethynyl-3-pyridyl-substituted subphthalocyanine 10 was obtained by standard Sonogashira cross-coupling reaction from triiodo SubPc 9 in 70% yield. Cage compound 8 was quantitatively self-assembled (Scheme 2) by mixing stoichiometric amounts of SubPc 10 and the palladium(II) complex (en)Pd(NO3)2 in a 1:1:1 mixture of water/methanol/acetonitrile followed by anion exchange with an excess of ammonium hexafluorophophate. The fullerene complex was prepared by adding 5 equivalents of C60 to a solution of capsule 8 in d6-acetone. 13C NMR spectrum of this mixture showed a broadening of the signals corresponding to the M3L2 cage 8 and the appearance of a peak at 140.1 ppm attributed to C60. In sharp contrast, 13C NMR of pure C60 in the same conditions does not show any signal since fullerene is not soluble enough in acetone (1 mg ml−1) [13]. Further evidence for the encapsulation of C60 was obtained from electrospray mass spectrometry in the same solvent that showed peaks at m/z = 1154.8, 829.7, 634.5 and 505.5 corresponding to [[C60 ⊂ 8]·3PF6]3+, [[C60 ⊂ 8]·2PF6]4+, [[C60 ⊂ 8]·PF6]5+ and [C60 ⊂ 8]6+, respectively, that compare well with theoretical exact mass values at 1153.2, 828.7, 633.9 and 504.1, respectively. Thus, in this way, one metallosupramolecular capsule 8 can extract a C60 molecule from the solid state and stabilize it in between the two concave surfaces of the subphthalocyanine halves. In addition to the magnetic interactions between the two components as evidenced by NMR spectroscopy, further photophysical studies are underway in order to deepen our understanding of the communication between the two π-extended surfaces.
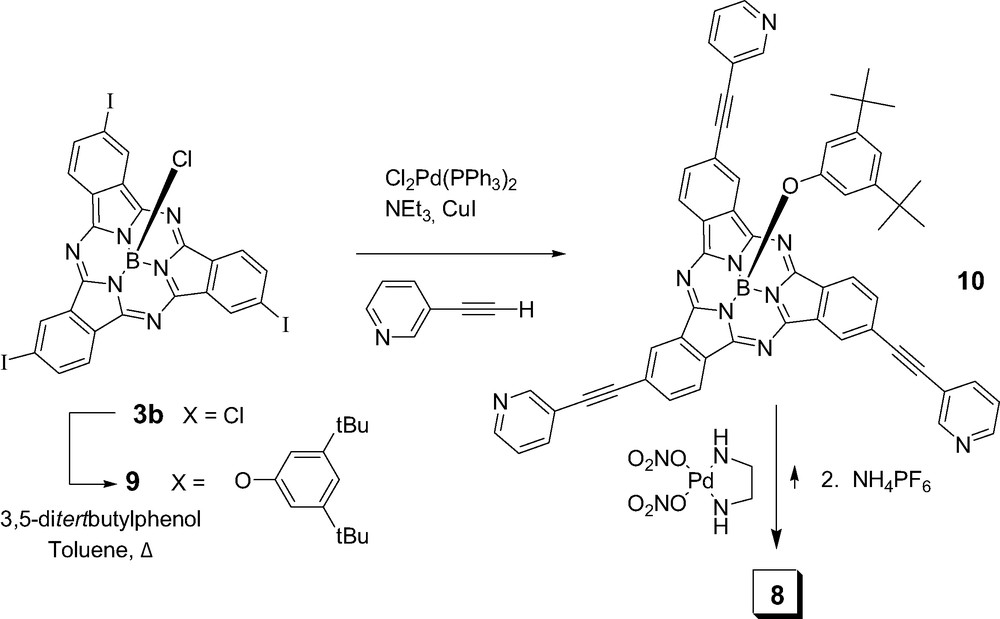
Synthesis of the M3L2 subphthalocyanine capsule 8.
5 Conclusions and outlook
Subphthalocyanines are shown to be extremely promising photo- and electroactive molecular building blocks for the construction of multicomponent assemblies in combination, for example, with C60 moieties. Photoinduced energy and electron transfer events take place in this kind of SubPc–C60 dyads following initial photoexcitation of the SubPc chromophore. On the other hand, they also represent ideal non-planar aromatic systems in which both convex and concave faces are accessible following covalent and supramolecular strategies.
This chemically versatile macrocycle is only beginning to emerge as a relevant molecule in the fields of photophysics, supramolecular chemistry and non-linear optics [14]. We are certain that there is still plenty of room for many useful applications of subphthalocyanines and subphthalocyanine derivatives.