

1 Introduction
While hundreds of ligands promote asymmetric additions to palladium bound π-allyls [1,2], the analogous asymmetric nickel chemistry is limited to a handful of papers [3]. The palladium processes are known to proceed by attack of soft stabilised nucleophiles (often malonate anions) exo to the allyl (A, Scheme 1). The nature of intermediate A dictates that bidentate ligands and symmetrical π-allyls will offer distinct advantages (the combination of P–N chelates and symmetrical allyls is particularly effective) [2]. Conversely, the nickel chemistry is normally proposed to proceed via endo migration of a Ni-bound nucleophile (normally an alkyl or other organogroup) to the allyl (B, Scheme 1) [3]. Intermediate B requires the presence of a monodentate chiral ligand for the allyl ligand to π-coordinate in simple square planar geometries. We speculated that under conditions where Nu is ‘small’ (e.g. a methyl group) and L is ‘large’ (i.e. a large chiral ligand) that the nickel chemistry might afford a new approach to substitution reactions of unsymmetrical π-allyl species (for which far fewer general approaches exist, particularly with non-malonate nucleophiles) [4] provided appropriate matching between the ligand and the derived nickel π-allyl could be attained.
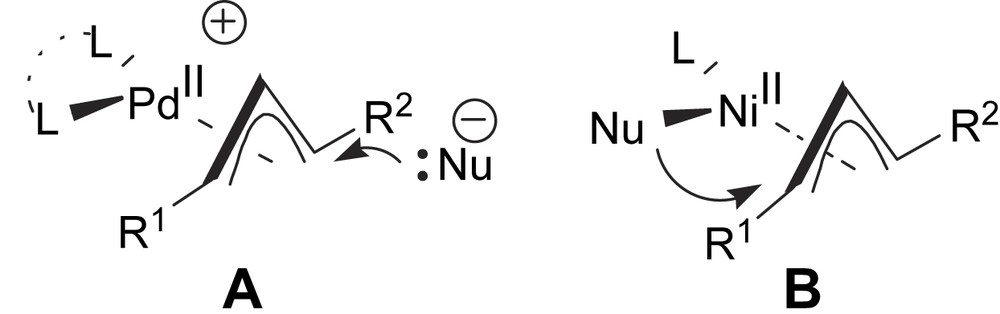
Comparison of Pd and Ni-catalysed allylation reactions.
2 Ligand selection and synthesis
We speculated that the recently introduced Ferrophite class of chiral ligands [5] might prove appropriate in attempting to define an effective asymmetric allylation reaction based on intermediate B whereby one of the four possible stereoisomers might be favoured. A small library of these compounds was selected for screening purposes (Scheme 2). Ferrophites L1–L7 were prepared either by our literature route or minor modifications thereof [5]. Installation of the phosphite unit used identical procedures to the synthesis of L1. Full preparative details of the new ligands L3 and L7 are given in the Section 5. To increase the diversity of the library and to compare its performance against known ligands of ‘privileged’ structure the ferrocenyl phosphine of Kagan (L8) [6] was included along with commercially available (R)-BINAP and (R,S)-Josiphos.
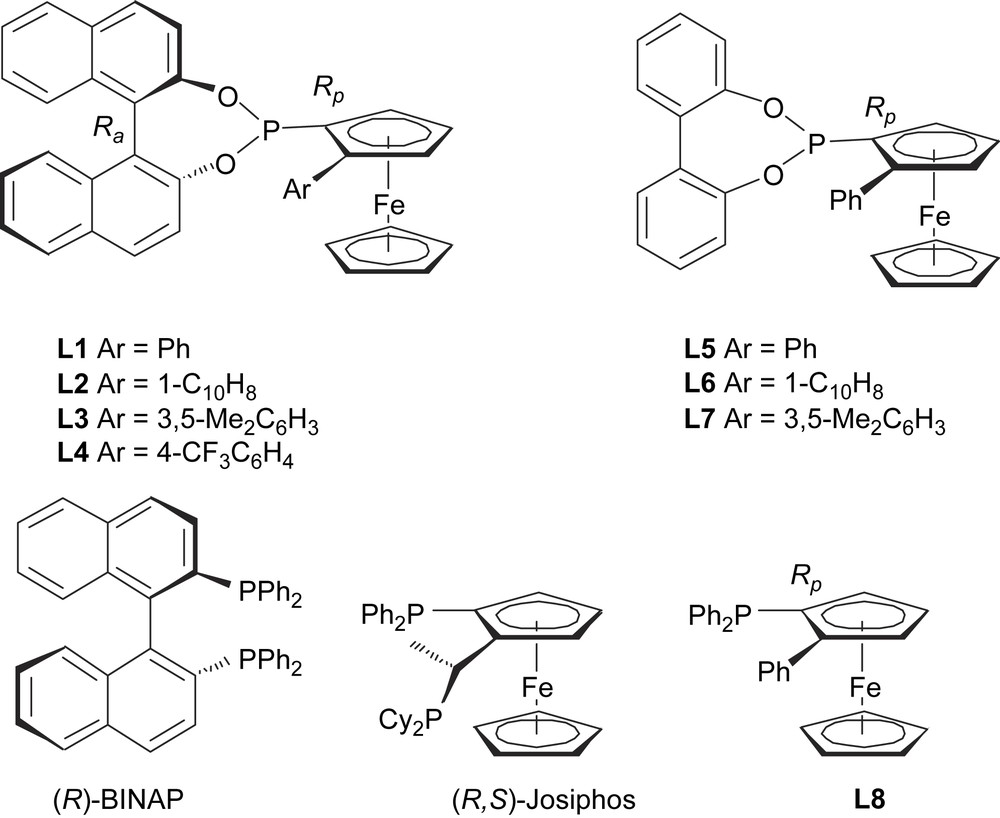
Preliminary ligand screening library.
3 Catalysis results and mechanistic inferences
We selected the conversion of the allylic chloride (1a) and acetate (1b) as representative non-symmetrical allylic electrophiles for two reasons: firstly, these species are readily attained through simple Baylis–Hillman chemistry [7], and secondly, a reliable chiral GC assay for the conversion yield and ee determination of product 2 was already available [8] (Scheme 3). The ether 1c was selected to allow direct comparison with the work of Nobuyoshi and RajanBabu [3a]. Preliminary results revealed that, in contrast to our previously investigated copper chemistry [8], formation of regioisomeric 3 competed with the desired γ product 2. In the absence of any added ligand, 3 was formed as essentially the sole AlMe3 displacement product (α/γ attack > 60:1). This fortunately enabled a GC assay for 3 to be developed as mixtures of 2/3 co-eluted under normal flash chromatography. The nickel-catalysed reaction required only low loadings [2 mol% Ni(acac)2] but was relatively slow, around 24 h was required at ambient temperature (conditions A) for complete conversion. Most asymmetric screenings were carried out for 3 days at 10 °C to maximise the enantioselectivities realised (conditions B) leading to somewhat lower conversions.
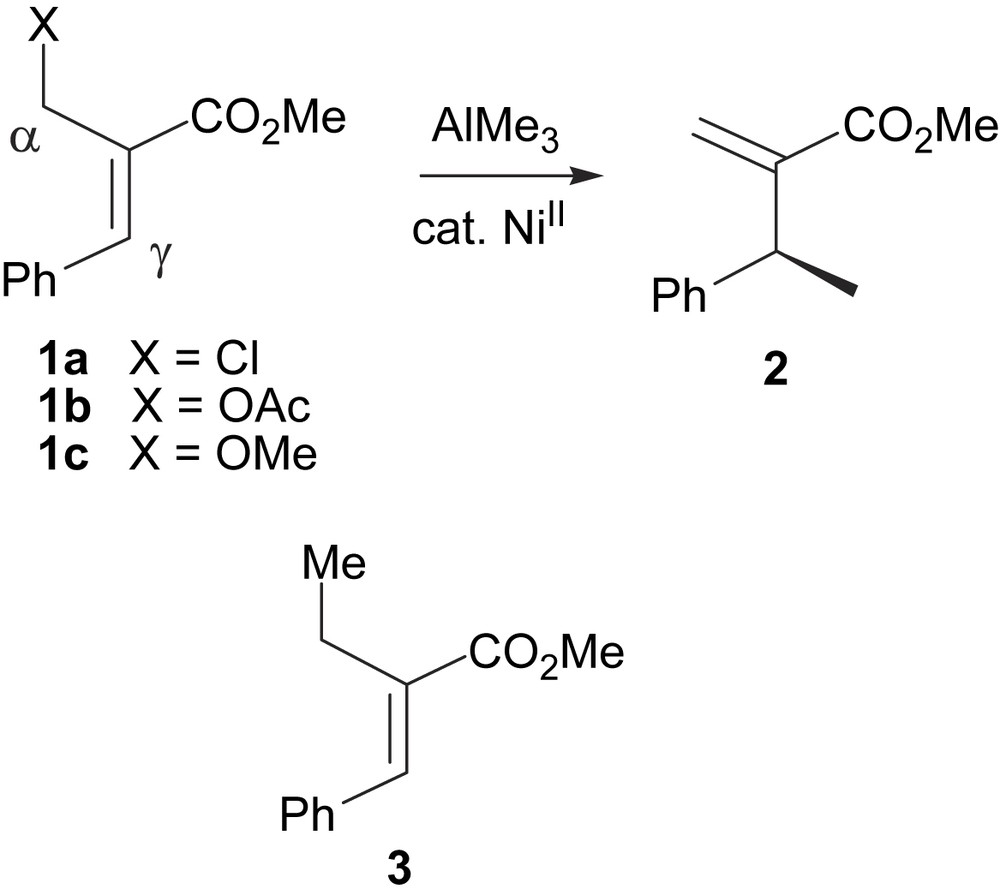
Methylation of Baylis–Hillman derived electrophiles.
As predicted, based on the structure of intermediate B, the chelate ligands BINAP and Josiphos were ineffective (runs 2 and 3). Utilisation of the monodentate Ferrophite ligand family in contrast delivered the desired product 2 with synthetically viable levels of enantioselectivities (up to 94%), though α/γ regioselectivity remained low in the majority of cases.
Comparison of the substrates 1a–c under catalysis by the biphenol ligand L5 was informative (runs 11–13). All three provided 2 with identical enantioselectivity (71%, S) consistent with the formation of a common π-allyl intermediate. The differing α/γ regiochemical ratios observed suggest that the rate of oxidative displacement at the α,γ-related termini of 1 is responsible for the observed regiochemical behaviour. This working model requires that the overall rate of the oxidative addition (and related processes) leading to the isomers C, ent-C, D and ent-D must be similar to the rate of expulsion of the products 2 and 3 from the catalysts. The implication that a mixture of four stereoisomeric π-allyl complexes are present is currently under both NMR and computational investigations. The presence of D/ent-D in the reaction mixture predicts that the bulk of the ligand should effect the regiochemistry (product 3 being achiral) independently of enantioselective events leading to 2. This notion is supported when using L2 and L6 whereby a far superior regioselectivity is observed for substrate 1b over 1a. Our working model for the process is that as the bulk of the ligand is increased, the formation of one of the less hindered π-allyls C/ent-C is favoured over diastereomeric D/ent-D (Scheme 4). However, as formation of C/ent-C requires γ attack of the LNi0 nucleophile on 1 during the oxidative addition if the steric demands of the ligand become too great then the overall reaction rate slows down. These simple ideas are supported by the data of Table 1 in the case of bulky L2 (low α/γ ratio) and conversely in the absence of any steric bulk (no added Ferrophite, L = solvent, maximum α/γ). The complete situation is complicated as we do not know if the shown anti allyl configuration is preferred and the conformation of the Ferrophite in the enantioselective reductive elimination also remains unknown. At present this creates anomalies in our analysis. For example, increasing the bulk of the aryl group in L1 from Ph to 3,5-C6H3Me2 (L3) produced little effect suggesting that the back of these substituents points into ‘free space’. However, closely related L7 did apparently favour formation of C/ent-C leading to improved regio but not enantioselectivity. Having shown that the general approach is valid searching for key π-allyl/ligand interactions by calculative methods will be profitable and this will be discussed in the future.
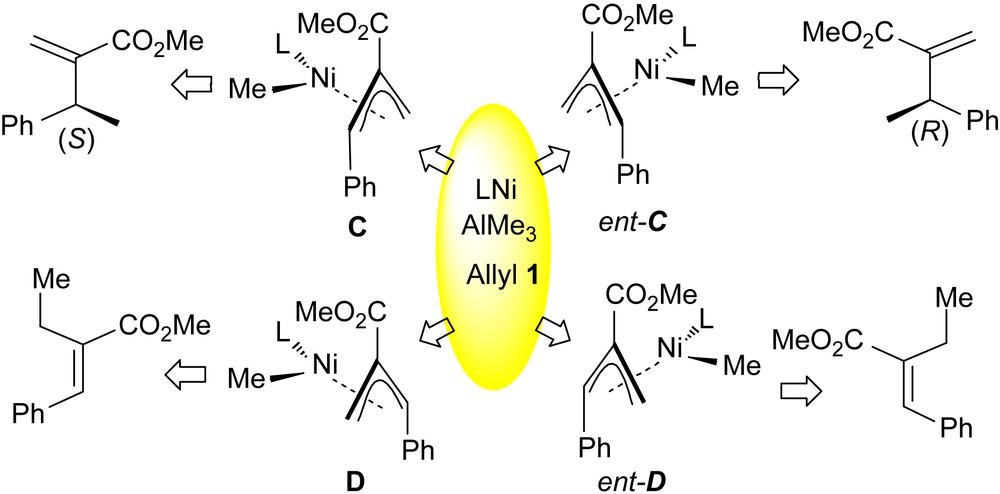
Working model for the observed selectivities.
Asymmetric substitution reactions of allylic electrophiles 1a
| Run | 1 (X) | Ligand | Method | Time/days | Conv./% | Yield 2 + 3/% | α/γ | ee 2 |
| 1 | 1a (Cl) | None | A | 3 | >99 | 63 | >60 | – |
| 2 | 1a (Cl) | BINAP | B | 2 | 26 | 8 | 1.65 | 6 (R) |
| 3 | 1a (Cl) | Josiphos | B | 2 | 37 | 16 | 0.33 | 4 (R) |
| 4 | 1a (Cl) | L1 | A | 1 | >99 | 82 | 2.04 | 58 (S) |
| 5 | 1a (Cl) | L2 | A | 1 | 61 | 36 | 0.50 | 84 (S) |
| 6 | 1a (Cl) | L2 | B | 3 | 52 | 30 | 0.58 | 92 (S) |
| 7 | 1a (Cl) | L2 | Bb | 3 | >99 | 41 | 1.20 | 94 (S) |
| 8 | 1b (OAc) | L2 | B | 3 | 48c | 33 | 0.22 | 94 (S) |
| 9 | 1a (Cl) | L3 | B | 3 | >99 | 71 | 1.96 | 52 (S) |
| 10 | 1a (Cl) | L4 | B | 3 | >99 | 77 | 1.14 | 73 (S) |
| 11 | 1a (Cl) | L5 | B | 3 | 82 | 73 | 0.52 | 71 (S) |
| 12 | 1b (OAc) | L5 | B | 2 | 50c | 26 | 0.30 | 71 (S) |
| 13 | 1c (OMe) | L5 | B | 3 | 31 | 24 | 0.85 | 71 (S) |
| 14 | 1a (Cl) | L6 | B | 3 | 98 | 43 | 0.50 | 91 (S) |
| 15 | 1b (OAc) | L6 | B | 3 | 72c | 19 | 0.16 | 93 (S) |
| 16 | 1a (Cl) | L7 | B | 3 | 70 | 56 | 0.75 | 49 (S) |
| 17 | 1a (Cl) | L8 | B | 3 | >99 | 66 | 4.08 | 63 (S) |
a Reactions carried out on 0.25 mmol 1 with 0.02/0.04/2 equiv. Ni(acac)2/ligand/AlMe3 in THF (3.0 mL). Conversions, yields and enantioselectivities by GC.
b DABAL–Me3 1.5 equiv. used.
c Conversion of acetate 1b determined by 1H NMR.
In addition to this, use of DABAL–Me3 [9] (an air-stable form of AlMe3) gave increased levels of enantioselectivity, but a loss of α/γ regioselectivity (run 7). This suggests that the stabilising amine (DABCO) remains bound to AlMe3 during both the transmetalation and enantioselective events.
4 Conclusions
We have been able to show a rare example of an enantioselective nickel-catalysed allylation reaction for non-symmetrical allylic electrophiles derived from Baylis–Hillman products. The enantioselectivities realised are significant (49–94%) and partial control of the regiochemistry of the addition can be attained through the ligand (up to α/γ = 0.16). In the absence of added ligand the reaction is regiospecific leading to the achiral substitution product. Although the reaction is viable at low catalyst loadings (2 mol% Ni/4 mol% ligand) its rate is insufficient and new ligand/condition combinations should be sought to significantly improve the present situation.
5 Experimental
5.1 General experimental
Reactions were carried out under conditions and equipment previously described [5]. Ligands L1, L2, L4–L6 and L8 were prepared by literature procedures [5,6]. BINAP and Josiphos were commercial samples. Compounds 1a and 1b were prepared according to literature procedures [7,10].
5.2 Preparation of the new ligands
5.2.1 (Rp,Sc)-2-(Tolyl-4-sulfinyl)-1-(3,5methylphenyl)ferrocene [CpFe{η5-1,2-C5H3(3,5-CH3C6H3)[S(O)-4-Tol]}] (precursor to L3 and L7)
To a mixture of [CpFe{η5-1,2-C5H3[B(OH)2][S(O)-4-Tol]}] (1.78 g, 4.82 mmol), palladium(dppf)dichloride ·CH2Cl2 (0.39 g, 0.48 mmol), 5-bromo-m-xylene (0.98 mL, 7.24 mmol) and toluene (80 mL) under argon was added sodium hydroxide (2 M, 4.7 mL, 9.41 mmol). The solution was heated at reflux for 4 h, then cooled, concentrated under reduced pressure and purified by column chromatography on silica gel (pentane/EtOAc/CH2Cl2, 7:2:1) to give the title compound as an orange solid. Yield: 1.255 g (61%). M.P. 202–204 °C. 1H NMR (400.1 MHz, CDCl3): δ 7.74 (d, 2H, J = 8.4 Hz, C6H4Me), 7.38 (s, 2H, C6H3Me2), 7.33 (d, 2H, J = 7.6 Hz, C6H4Me), 6.93 (s, 1H, C6H3Me2), 4.68–4.67 (m, 1H, C5H3), 4.38 (t, 1H, J = 2.4 Hz, C5H3), 4.13 (s, 5H, C5H5), 4.06–4.05 (m, 1H, C5H3), 2.44 (s, 3H C6H4Me), 2.36 (s, 6H, C6H3Me2) ppm. 13C{1H}NMR (100.1 MHz, CDCl3): δ 141.3 (C6H3Me2), 140.1 (C6H3Me2), 137.6 (C6H3Me2), 135.5 (C6H3Me2), 129.2 (C6H4Me), 129.1 (C6H3Me2), 127.6 (C6H3Me2), 125.7 (C6H4Me), 92.3 (C5H3), 90.3 (C5H3), 72.1 (C5H3), 71.1 (C5H5), 69.2 (C5H3), 68.9 (C5H3), 21.5 (C6H4Me), 21.4 (C6H3Me2) ppm. IR (CH2Cl2): ν = 2921, 1601, 1493, 1108, 1084, 1035, 1015, 853, 828 cm−1. [Found (HRMS, ES): MH+ 429.0973. C25H25OSFe requires M 429.0970.]
5.2.2 (Rp,Ra)-2-(3,5-Dioxa-4-phosphacyclohepta[2,1-a;3,4-a′]dinaphthalen-4-yl)-1-m-xyleneferrocene [CpFe{η5-1,2-C5H3(3,5-CH3C6H3)[P(O2C20H12)]}] (L3)
A solution of sulfoxide precursor (0.45 g, 1.05 mmol) in THF (10 mL) was cooled to −78 °C under argon. To the solution was added tBuLi (1.7 M in hexanes, 0.68 mL, 1.16 mmol) which caused the solution to darken. After 5 min binaphthyl phosphite was added (0.43 g, 1.05 mmol) as a THF solution (5 mL), causing the solution to lighten in colour. After 5 min, water (1 mL) was added to quench the reaction and was warmed quickly to room temperature. The reaction mixture was extracted with ether and the organic layer dried over MgSO4, concentrated under reduced pressure and purified further by column chromatography on silica gel (pentane/Et2O, 4:1) to give the title compound as an orange solid. Yield: 0.31 g (54%) [α]D = −230 (c = 1.00, CH2Cl2). 31P{H}NMR (202.5 MHz, C6D6): δ 187.9 ppm. 1H NMR (500.1 MHz, C6D6): δ 7.73–7.70 (m, 2H, Ar), 7.68 (s, 2H, Ar), 7.64 (d, 1H, J = 8.5 Hz, Ar), 7.62 (d, 1H, J = 8.0 Hz, Ar), 7.55 (d, 1H, J = 8.5 Hz, Ar), 7.52 (d, 1H, J = 8.5 Hz, Ar), 7.48 (d, 1H, J = 8.5 Hz, Ar), 7.21–7.14 (m, 2H, Ar), 7.10 (d, 1H, J = 9.0 Hz, Ar), 7.02–6.97 (m, 2H, Ar), 6.80 (s, 1H, Ar), 4.59–4.58 (m, 1H, C5H3), 4.12 (s, 5H, C5H5), 3.92–3.89 (m, 2H, C5H3), 2.29 (s, 6H, C6H3Me2) ppm. 13C{H}NMR (100.6 MHz, C6D6): δ 150.2, 150.1, 138.0, 138.0, 133.7, 133.3, 132.1, 131.5, 131.0, 129.8, 129.2, 128.8, 127.4, 127.2, 126.7, 126.5, 125.7, 125.6, 125.1, 124.9, 124.4, 123.1, 122.2, 94.33 (d, J = 25 Hz), 74.1, 74.1, 71.1, 71.0, 21.5 ppm. IR (CH2Cl2): ν = 2917, 2855, 1601, 1589, 1510, 1330, 1106, 1070, 950, 851, 827 cm−1. [Found (HRMS, ES): MH+ 605.1318. C38H30O3PFe requires M 605.1327.]
5.2.3 (Rp,Ra)-2-(3,5-Dioxa-4-phosphacyclohepta[2,1-a;3,4-a′]biphenalen-4-yl)-1-m-xyleneferrocene [CpFe{η5-1,2-C5H3(3,5-CH3C6H3)[P(O2C12H8)]}] (L7)
A solution of sulfoxide precusor (0.54 g, 1.25 mmol) in THF (15 mL) was cooled to −78 °C under argon. To the solution was added tBuLi (1.7 M in hexanes, 0.81 mL, 1.38 mmol) which caused the solution to darken. After 5 min biphenyl phosphite was added (0.39 g, 1.25 mmol) as a THF solution (5 mL), causing the solution to lighten in colour. After 5 min, water (1 mL) was added to quench the reaction, and was warmed quickly to room temperature. The reaction mixture was extracted with ether and the organic layer dried over MgSO4, concentrated under reduced pressure and purified further by column chromatography on silica gel (pentane/Et2O, 4:1) to give the title compound as an orange solid. Yield: 0.05 g (8%) [α]D = 22.6 (c = 1.00, CH2Cl2). 31P{H}NMR (202.5 MHz, C6D6): δ 190.1 ppm. 1H NMR (500.1 MHz, C6D6): δ 7.63 (s, 2H, Ar), 7.28 (dd, 2H, J = 7.5, 1.5 Hz, Ar), 7.21 (d, 1H, J = 7.5 Hz, Ar), 7.12 (dt, 1H, J = 7.5, 1.5 Hz, Ar), 7.03 (t, 1H, J = 7.5 Hz, Ar), 6.96 (dt, 1H, J = 7.5, 1.0 Hz, Ar), 6.91 (dt, 1H, J = 8.0, 2.0 Hz, Ar), 6.83 (d, 1H, J = 8.0 Hz, Ar), 6.78 (s, 1H, Ar), 4.60–4.58 (m, 1H, C5H3), 4.25–4.24 (m, 1H, C5H3), 4.14 (s, 5H, C5H5), 4.08 (t, 1H, J = 2.5 Hz, C5H3), 2.26 (s, 6H, C6H3Me2) ppm. 13C{H}NMR (100.6 MHz, C6D6): δ 152.7, 152.7, 152.1, 152.0, 138.0, 137.9, 133.3, 133.2, 132.2, 132.2, 130.1, 130.0, 129.6, 129.2, 129.1, 125.3, 124.7, 122.8, 122.4, 122.4, 94.36 (d, J = 25 Hz), 74.4, 74.0, 74.0, 71.2, 71.1, 71.1 ppm. IR (CH2Cl2): ν = 3557, 2922, 1602, 1500, 1332, 1196, 1098, 1039, 1009, 853 cm−1. [Found (HRMS, ES): MNa+ 523.1111. C30H28O3PFeNa requires M 523.1120.]
5.2.4 (E)-Methyl 2-(methoxymethyl)-3-phenylacrylate (1c) [11]
To a stirred solution of NaH (60% dispersion in mineral oil) (0.08 g, 2 mmol) in dry THF (10 mL) at 0 °C under an argon atmosphere was added a solution of precursor alcohol (E)-PhCHC(CH2OH)(CO2Me) (0.39 g; 2 mmol) in dry THF (5 mL) over 1 h. To the reaction mixture MeI (150 μL; 2.4 mmol) was added and the mixture stirred for a further 18 h at room temperature. Quenching the reaction with sat. NH4Cl (aq) (20 mL) was followed by extraction with Et2O (50 mL). The combined organic layers were washed with brine (50 mL) and dried over MgSO4. Purification by flash chromatography on silica gel (pentane/Et2O, 4:1) gave the title compound as a colourless oil. Yield: 0.24 g (58%) 1H NMR (270 MHz, CDCl3): δ 7.68 (s, 2H, Ar), 7.95 (s, 1H, CH), 7.51–7.40 (m, 3H, Ar), 4.25 (s, 2H, CH2OMe), 3.86 (s, 3H, C(O)OMe), 3.45 (s, 3H, OMe) ppm. 13C{H}NMR (100.6 MHz, CDCl3): δ 168.1, 144.8, 134.7, 130.1, 129.9, 129.4, 128.6, 66.6, 58.3, 52.2 ppm. [Found (HRMS, ES): MNa+ 229.0829. C12H14O3Na requires M 229.0835.]
5.3 General procedure
5.3.1 Nickel-catalysed asymmetric alkylation of Baylis–Hillman derived electrophiles with AlMe3
To a flame-dried Schlenk tube under argon charged with ligand (4 mol %) and Ni(acac)2 (1.2 mg, 2 mol%) anhydrous THF (3 mL) was added and stirred at −10 °C for 5 min. To this the neat allylic chloride or acetate (0.25 mmol) was added and the mixture stirred at −10 °C for 10 min after which time AlMe3 (2 M solution in heptanes, 0.5 mmol, 0.25 mL) was added dropwise. The reaction mixture was warmed to 10 °C, after 3 days at this temperature the yellow reaction was quenched with 2 M HCl (2 mL) followed by addition of the GC internal standard (pentadecane, 25 μL) and extraction with Et2O (2 × 3 mL). The organic phase was separated and filtered through a plug of silica. A 1 mL sample was removed for GC analysis.
5.3.2 (±)-Methyl 2-methylene-3-phenylbutanoate (2)
Pure compound prepared according to literature procedure [7] gave colourless oil. 1H NMR (270 MHz, CDCl3): δ 7.30–7.16 (m, 5H, Ar), 6.28 (s, 1H, CH2α), 5.61 (s, 1H, CH2β), 4.03 (q, 1H, J = 7.2 Hz, CHMe), 3.67 (s, 3H, OMe), 1.42 (d, 3H, J = 7.2 Hz, Me) ppm. 13C{H}NMR (100.6 MHz, CDCl3): δ 167.6, 145.0, 144.5, 128.6, 127.6, 126.5, 124.0, 52.0, 40.7, 20.9 ppm. IR (CHCl3): ν = 2969m, 1721s, 1627, 1437, 1149, 947 cm−1. [Found (HRMS, EI): M+ 190.0998. C12H14O2 requires M 190.0998.] The GC assay: 25m 2,6-O-dimethyl,3-O-pentyl)-γ-cyclodextrin silica column, 120 °C (30 min): 10 °C/min: 200 °C (20 min), He carrier gas, 12 psi, (S): 14.8 min. (R): 15.2 min. Anal. Calcd. for C12H14O2: C 75.76, H 7.42%; found: C 75.75, H 7.37%.
5.3.3 (E)-Methyl 2-benzylidenebutanoate (3) [12]
Prepared according to general procedure A in absence of ligand. Purification by flash chromatography on silica gel (hexane/Et2O, 9:1) gave the title compound as a colourless oil. Isolated yield: 20 mg (42%) 63% by GC. 1H NMR (270 MHz, CDCl3): δ 7.66 (s, 1H, CH), 7.40–7.35 (m, 5H, Ar), 3.83 (s, 3H, OMe), 2.57 (q, 2H, J = 7.6 Hz, CH2Me), 1.18 (t, 3H, J = 7.6 Hz, Me) ppm. 13C{H}NMR (100.6 MHz, CDCl3): δ 168.8, 138.6, 135.8, 134.8, 129.2, 128.3, 128.0, 51.9, 20.8, 13.9 ppm. IR (CHCl3): ν = 3027, 2952, 2877, 1698, 1628, 1495, 1435, 1312, 1284, 1238, 1204, 1130, 1045, 810 cm−1. [Found (HRMS, EI): MNa+ 213.0898. C12H14O2Na requires MNa 213.0897.] The GC assay: 25m 2,6-O-dimethyl,3-O-pentyl)-γ-cyclodextrin silica column, 120 °C (30 min): 10 °C/min: 200 °C (20 min), 24.9 min.
Acknowledgements
We are grateful to the EU Commission for support through contract (LigBank). One of us (AN) thanks GlaxoSmithKline for provision of a studentship.