

1 Introduction
Homotypic interactions between biopolymers such as nucleic acid–nucleic acid and protein–protein are the basis for bio-recognition and cell functioning, as they mediate such processes as transcription, signal transduction and cell membrane formation. It is well known that cell surface carbohydrates play a role in cell–cell adhesion and communication [1]. Several models for cell interactions based on carbohydrates have been proposed. In most of them, the carbohydrate binds to selectins, galectins, and other carbohydrate-binding proteins [2]. Recent advances in glycobiology revealed the existence of biologically significant carbohydrate–carbohydrate interactions, and this type of interaction could have a general, fundamental character for cell biology [3]. A typical example is the report of Hakomori, who proposed that carbohydrate–carbohydrate interaction is responsible for the initial step of cell adhesion [4]. Embryogenesis, metastasis, and other proliferation processes are, according to Hakomori, mediated by carbohydrate–carbohydrate interactions [4]. One of the structures involved in this novel mechanism is the Lewisx (Lex) trisaccharide determinant (Galβ1 → 4[Fucα1 → 3]GlcNAcβ1). The Lewisx antigen – previously defined as the Stage-Specific Embryonic Antigen 1 (SSEA-1) – is found in a wide variety of systems such as human cancers, pre-implantation mouse embryos, embryonic carcinoma cells, and human erythrocytes [5].
The interaction between Lex and Lex was found to be homotypic and mediated by the presence of divalent cations such as Ca2+ [6,7]. Recently, the Lex–Lex interaction has increasingly been studied, using a variety of techniques including nuclear magnetic resonance (NMR) spectroscopy [8], mass spectrometry (MS) [9], vesicle adhesion [10], atomic force microscopy (AFM) [11], and surface plasmon resonance (SPR) spectroscopy [12]. Rat basophilic leukaemia cells pre-incubated with purified Lex containing glycosphingolipids have also been used as a model [13]. Another model system, “Glycosylated Foldamer”, has been demonstrated very recently to study the carbohydrate–carbohydrate interaction in terms of individual carbohydrate motifs [14]. To demonstrate the homotypic characterization of this kind of interactions, we recently performed an experiment using the vesicle technique [15]. In this study, the Lex was replaced by Lea, in which the galactose and fucose are permutated relative to the Lex on one vesicle surface. The weak adhesion energy obtained for the Lex–Lea pair showed clearly that the permutation of the fucose and galactose residues in the trisaccharide headgroup effectively prevents specific adhesion [15].
To date, however, the molecular detail of these weak, Ca2+-dependent interactions remains unclear, necessitating the new models to probe the nature of this phenomenon in terms of key role of the different hydroxyl groups on Lex trisaccharide involved in the Lex–Lex interaction. Our objective has been to prepare a series of Lex pentasaccharides in which one of the eight hydroxyl groups of Lex trisaccharide is replaced by an hydrogen atom (Fig. 1) and to test the induced adhesion by interaction of these derivatives, in order to gain an insight into the functions played by the hydroxyl groups of the Lex trisaccharide.

Lewisx pentasaccharide.
Taking the cost of different sugar residues of Lewisx pentasaccharide into account, the synthetic work was started from the chemical modification of the galactose residue. After successful synthesis of 3d-deoxy Lewisx pentasaccharide [16], we describe here the first total synthesis of 4d-deoxy Lewisx pentasaccharide 1 (Fig. 2), which is a useful tool for carbohydrate–carbohydrate interaction study.
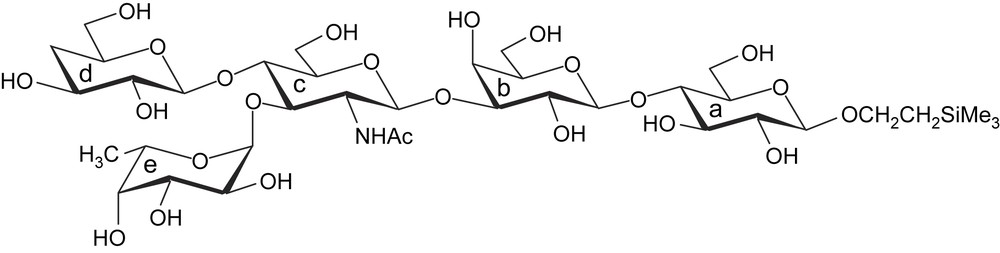
4d-Deoxy Lewisx pentasaccharide 1.
Classic strategy for oligosaccharide assembly is involved in the manipulation of the protecting groups between each glycosylation step. Such a manipulation is a consequence of increased linearity and inefficiency of oligosaccharide assembly. In order to increase assembly efficiency of complex oligosaccharides by avoiding various unnecessary manipulations of each glycosylation step, an intelligent strategy is to use highly stereo- and regioselective glycosylation of partially or unprotected acceptors [17] based on differences in reactivity of the hydroxyl groups followed by analysis of the structures using modern 2D NMR techniques. Another useful strategy is to employ two-directional glycosylation, which exploits both the differences in the reactivities of an anomeric leaving group and the subtle control of nucleophilicities of sugar hydroxyl groups [18]. The major purpose of these strategies is to overcome the traditional, tedious multi-step protection/deprotection schemes and provide an efficient way for the complex oligosaccharide synthesis. These strategies have been successfully demonstrated in our first synthesis of 3d-deoxy Lewisx pentasaccharide [16] and were proved to be effective in our following syntheses of disaccharide and pentasaccharide.
2 Results and discussion
O-(2,3,6-Tri-O-benzoyl-4-deoxy-α-d-xylo-hexopyranosyl)trichloroacetimidate (4) was chosen as galactosyl donor to construct a lactosamine building block for the synthesis of the pentasaccharide 1. This imidate per se was prepared from phenyl 2,3,6-tri-O-benzoyl-4-deoxy-1-thio-β-d-xylo-hexopyranoside 2 [19]. Thus treatment of 2 with BF3·Et2O in the presence of HgO gave the hemiacetal 3 as a mixture of α/β isomers in 88% yield, which were not separated and further characterized. Compound 3 was then treated with trichloroacetonitrile in the presence of 1,8-diazabicyclo[5,4,0]undec-7-ene (DBU) as a base to provide the trichloroacetimidate 4 in 77% yield, as outlined in Scheme 1.
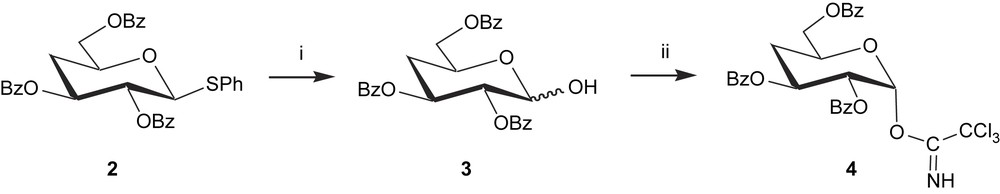
Reagents and conditions: (i) BF3·Et2O, HgO, H2O, THF, RT, 1 h, 88%; (ii) Cl3CCN, DBU, CH2Cl2, 0 °C, 1 h, 85%.
The NMR spectra showed that only the α-trichloroacetimidate was formed on the basis of the H-1, H-2 coupling constant (J1,2 = 3.7 Hz). This is because an axial trichloroacetimidate is the thermodynamically more stable isomer [20]. However, a thermodynamically unstable equatorial imidate could also be occasionally isolated in a similar reaction [21].
As illustrated in Scheme 2, condensation of the trichloroacetimidate 4 with the previously reported diol 5 [22], according to the Schmidt glycosylation procedure [23], using TMSOTf as a promoter without affecting the SPh group of the acceptor 5, proceeded regioselectively, taking advantage of the “stereo hindrance effect”, to give the β1 → 4 linked disaccharide 6a in 80% yield; the by-product β1 → 3 linked disaccharide 6b was isolated as a minor product (9%). Such a selective behaviour of phenyl 6-O-benzyl-2-deoxy-2-phthalimido-1-thio-β-d-glucopyranoside 5 has previously been observed during its glycosylation with 2,3,4,6-tetra-O-benzoyl-α-d-galactopyranosyl bromide [24] or with O-(3-O-acetyl-2,6-di-O-benzoyl-3-deoxy-α-d-xylo-hexopyranosyl)trichloroacetimidate [16]. Another example for this type of regioselective glycosylation was reported for phenyl 2,3,4 tri-O-acetyl-6-O-benzyl-1-thio-β-d-galactopyranoside [25].
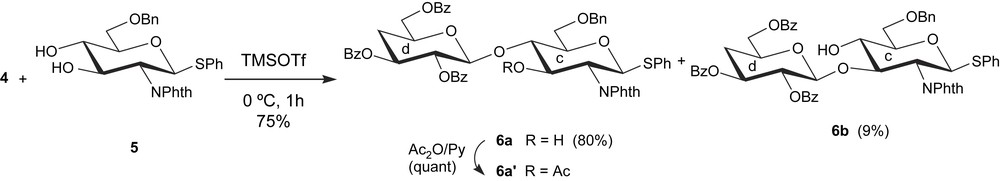
The 1H NMR spectrum of 6a showed the presence of H-3c of the glucosamine residue at δ 4.54 (ddd, J2c,3c = 10.3 Hz, J3c,4c = 8.3 Hz, J3c,OH = 1.0 Hz), indicating the position of the newly formed glycosidic linkage in the disaccharide 6a to be at OH-4 of the acceptor 5. This regioselectivity was further confirmed from the 1H NMR spectrum of 6a′ – obtained from 6a by acetylation – which revealed a deshielded signal for H-3c at 5.78 ppm (dd, J2c,3c = 10.1 Hz, J3c,4c = 9.2 Hz), confirming therefore the position of the new glycosidic linkage in 6a as being OH-4 of the diol 5. Its stereochemistry was determined to be desired β on the basis of the H-1d, H-2d coupling constant (J1d,2d = 7.9 Hz).
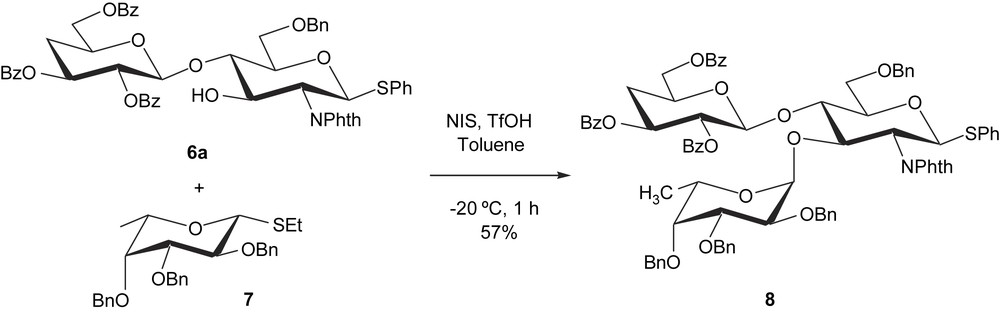
For construction of trisaccharide, the disaccharide 6a was fucosylated by the known ethyl 2,3,4-tri-O-benzyl-1-thio-β-l-fucopyranoside 7 [26] in toluene using N-iodosuccinimide (NIS)–trifluoromethanesulfonic acid (TfOH) as catalysts. The chemical yield of this glycosylation was found to be temperature dependent; the best yield was obtained when the reaction was run at −20 °C for 1 h, giving the expected trisaccharide 8 in 57% yield (Scheme 3). The stereochemistry of the newly introduced glycosidic linkage was determined to be α on the basis of the low value of the Fuc H-1, H-2 coupling constant (J1e,2e = 3.5 Hz).
The next glycosylation of the diol 9 [27] with the donor 8 was performed under the similar conditions described above, using dichloromethane as solvent instead of toluene. Again the outcome of this reaction was proved to be temperature dependent. The glycosylation was started at −30 °C by addition of the promoters into the reaction mixture, the temperature was then slowly raised to room temperature, the desired pentasaccharide 10 was isolated in excellent yield (Scheme 4). The stereochemistry of the newly introduced linkage was determined to be β on the basis of the GlcN H-1, H-2 coupling constant (J1c,2c = 8.4 Hz). The regiochemistry of 10 was assigned from the 1H NMR spectrum of 10′ – obtained from 10 by acetylation – which revealed a deshielded signal for H-4b at 5.42 ppm (d, J3b,4b = 3.5 Hz, J4b,5b being close to zero), confirming therefore the position of the new glycosidic linkage in 10 as being OH-3b of the diol 9. In fact, the regioselectivity of 3-OH was usually observed for glycosylation of the 3,4-diol of the galactose moiety, especially using a large glycosyl donor [28,29].
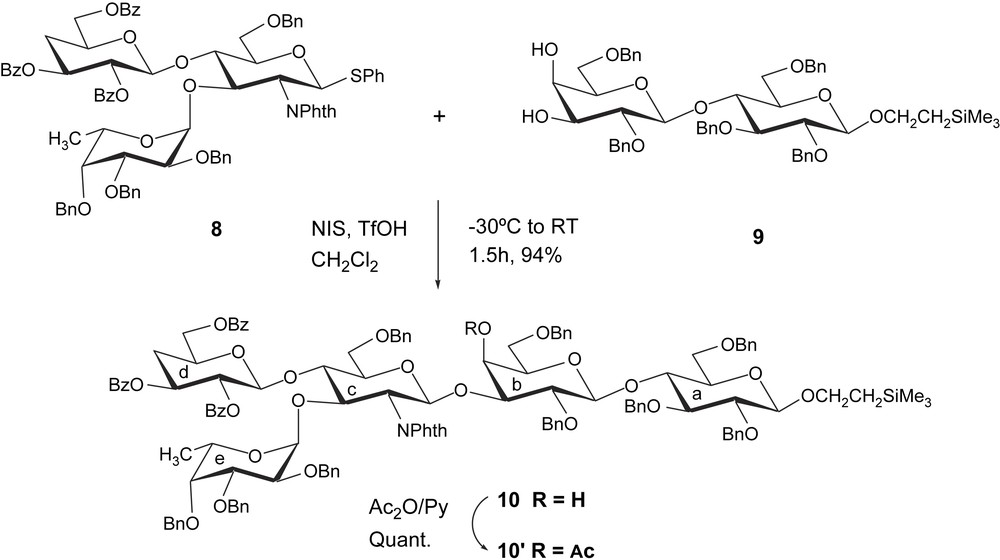
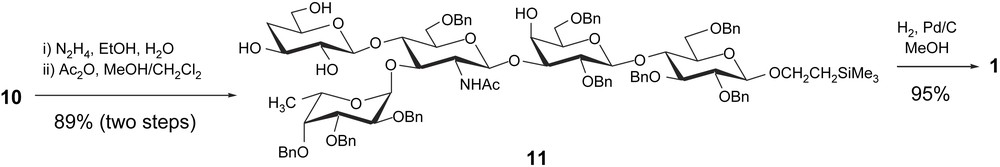
The final route to target oligosaccharide 1 is outlined in Scheme 5. Treatment of pentasaccharide 10 with hydrazine in boiling ethanol, followed by a selective N-acetylation, afforded the derivative 11 in 89% overall yield from 10. Catalytic hydrogenolysis of 11 in methanol for 13 h, followed by purification on Sephadex G25-150, provided the desired pentasaccharide 1 as a white amorphous solid in almost quantitative yield.
The structure and purity of the synthesized compounds were established by two-dimensional 1H–1H homonuclear correlation, 1H–13C heteronuclear correlation experiments and HR-FAB mass spectroscopy.
3 Experimental section
3.1 General methods
Optical rotations were measured at 20 ± 2 °C with a PerkinElmer Model 241 digital polarimeter, using a 10-cm, 1-mL cell. Fast Atom Bombardment (FAB) mass spectra were obtained with a JMS-700 spectrometer. 1H NMR spectra were recorded with a Bruker DRX 400 spectrometer at ambient temperature. Assignments were aided by COSY experiments. 13C NMR spectra were recorded at 100.6 MHz with a Bruker DRX 400 for solutions in CDCl3 or D2O. Reactions were monitored by thin-layer chromatography (TLC) on a precoated aluminium plate of silica gel 60 F254 (layer thickness 0.2 mm, E. Merck, Darmstadt, Germany) and detection by charring with sulfuric acid. Flash column chromatography was performed on silica gel 60 (230–400 mesh, E. Merck).
3.2 O-(2,3,6-Tri-O-benzoyl-4-deoxy-α-d-xylo-hexopyranosyl)trichloroacetimidate (4)
Trichloroacetonitrile (4.76 mL, 46.5 mmol) was added to a solution of 3 (1.67 g, 3.5 mmol) in 62 mL of dry dichloromethane at 0 °C, then DBU (0.62 mL, 4.0 mmol) was added dropwise. The mixture was stirred at 0 °C for 1 h. After concentration, the residue was purified by flash column chromatography (toluene–ethyl acetate–triethylamine 150:10:0.1) to afford 4 (1.83 g, 85%) as a white foam; Rf = 0.50 (toluene–ethyl acetate 9:1); [α]D +100 (c 0.45, chloroform); 1H NMR (400 MHz, CDCl3): δ 8.63 (s, 1H, NH), 8.10–7.30 (m, 15H, aromatic), 6.80 (d, 1H, J1,2 = 3.7 Hz, H-1), 5.96–5.88 (m, 1H, H-3), 5.64 (dd, 1H, J1,2 = 3.7 Hz, J2,3 = 10.1 Hz, H-2), 4.69–4.65 (m, 1H, H-5), 4.56–4.48 (m, 2H, H-6, H-6′), 2.65 (ddd, 1H, J3,4 = 4.9 Hz, J4,5 = 2.5 Hz, Jgem = 12.7 Hz, H-4), 2.08 (q, 1H, J = 12.7 Hz, H-4′); 13C NMR (100.6 MHz, CDCl3): δ 166.12, 165.76, 165.62 (3CO, Bz), 160.68 (CNH), 133.65–128.36 (aromatic CH), 94.28 (C-1), 70.90 (C-2), 68.47 (C-3), 68.27 (C-5), 65.50 (C-6), 32.56 (C-4); FAB-HRMS (m/z) Calcd for C29H24Cl3NO8Na (M + Na+): 644.0442. Found: 644.0428.
3.3 Phenyl (2,3,6-tri-O-benzoyl-4-deoxy-β-d-xylo-hexopyranosyl)-(1 → 4)-6-O-benzyl-2-deoxy-2-phthalimido-1-thio-β-d-glucopyranoside (6a) and phenyl (2,3,6-tri-O-benzoyl-4-deoxy-β-d-xylo-hexopyranosyl)-(1 → 3)-6-O-benzyl-2-deoxy-2-phthalimido-1-thio-β-d-glucopyranoside (6b)
A solution of 5 (0.79 g, 1.6 mmol, 1.0 equiv) and 4 (1.48 g, 2.4 mmol, 1.5 equiv) in 29 mL of dry dichloromethane was stirred with 4-Å powdered molecular sieves (2 g) for 40 min at room temperature under an argon atmosphere. Trimethylsilyl triflate (87.6 μL, 0.48 mmol, 0.3 equiv) was added dropwise at 0 °C. The mixture was stirred for 40 min and then filtered through Celite. The filtrate was washed with a saturated aqueous NaHCO3 and then with water, dried over MgSO4 and concentrated. The residue was submitted to flash column chromatography and eluted with toluene–ethyl acetate (9:1). The earlier fractions contained the compound 6a; on concentration, these fractions gave a white foam (1.21 g, 80%); Rf = 0.50 (toluene–ethyl acetate 4:1); [α]D +47 (c 1, chloroform); 1H NMR (400 MHz, CDCl3): δ 8.07–7.20 (m, 29H, aromatic), 5.60 (d, 1H, J1,2 = 10.3 Hz, H-1c), 5.50 (dd, 1H, J1,2 = 7.8 Hz, J2,3 = 9.8 Hz, H-2d), 5.43–5.39 (m, 1H, H-3d), 4.81 (d, 1H, J1,2 = 7.8 Hz, H-1d), 4.66 (dd, 1H, Jgem = 11.8 Hz, PhCH), 4.60 (d, 1H, J = 0.9 Hz, exch. D2O, OH-3c), 4.54 (ddd, 1H, J2,3 = 10.3 Hz, J3,4 = 8.3 Hz, J3,OH = 0.9 Hz, H-3c), 4.35 (t, 1H, J1,2 = 10.3 Hz, J2,3 = 10.3 Hz, H-2c), 4.23–4.08 (m, 4H, H-5d, H-6d, H-6d′, PhCH), 3.82–3.80 (m, 1H, H-4c), 3.71–3.67 (m, 1H, H-5c), 3.56–3.48 (m, 2H, H-6c, H-6c′), 2.47 (ddd, 1H, J3,4 = 5.1 Hz, J4,5 = 1.7 Hz, Jgem = 11.7 Hz, H-4d), 1.85 (q, 1H, J = 11.7 Hz, H-4d′); 13C NMR (100.6 MHz, CDCl3): δ 168.06, 167.44, 166.23, 165.71, 165.11 (5CO, NPhth, Bz), 138.25, 134.48, 132.19, 131.81, 131.66, 129.06, 129.02 (aromatic C), 134.48, 133.99, 133.93, 133.50, 133.37, 133.19, 132.40, 129.84, 129.83, 129.69, 128.73, 128.52, 128.42, 128.27, 127.70, 127.51, 127.36, 123.56, 123.58 (aromatic CH), 101.76 (C-1d), 83.29 (C-1c), 82.41 (C-4c), 77.80 (C-5c), 72.88 (C-6c), 72.28 (C-2d), 70.97 (C-3d), 70.89 (C-3c), 70.53 (C-5d), 68.18 (C-6d), 65.41 (PhCH2), 55.02 (C-2c), 32.31 (C-4d); FAB-HRMS (m/z) Calcd for C54H51N2O13S (M + NH4+): 967.3112. Found: 967.3116.
The later fractions contained 6b as a syrup (142 mg, 9.4%); Rf = 0.43 (toluene–ethyl acetate 4:1); [α]D +102 (c 1, chloroform); 1H NMR (400 MHz, CDCl3): δ 8.19–7.13 (m, 29H, aromatic), 5.43 (d, 1H, J1,2 = 10.4 Hz, H-1c), 5.38 (dd, 1H, J1,2 = 7.8 Hz, J2,3 = 9.8 Hz, H-2d), 5.34–5.28 (m, 1H, H-3d), 4.72 (d, 1H, J1,2 = 7.8 Hz, H-1d), 4.68–4.58 (m, 5H, H-3c, H-6d, OH-4c, PhCH2), 4.41 (t, 1H, J1,2 = 10.4 Hz, J2,3 = 10.4 Hz, H-2c), 4.33 (dd, 1H, Jgem = 12.0 Hz, J5,6 = 7.6 Hz, H-6d′), 4.20–4.14 (m, 1H, H-5d), 3.97 (d, 1H, J = 10.4 Hz, H-5c), 3.80–3.70 (m, 3H, H-4c, H-6c, H-6c′), 2.41 (ddd, 1H, J3,4 = 5.2 Hz, J4,5 = 1.8 Hz, Jgem = 12.8 Hz, H-4d), 1.84 (q, 1H, J = 12.8 Hz, H-4d′); 13C NMR (100.6 MHz, CDCl3): δ 168.53, 166.70, 166.13, 165.63, 164.85 (5CO, NPhth, Bz), 138.48, 132.68, 130.79, 130.71, 129.21, 128.85, 128.76 (aromatic C), 133.93, 133.56, 133.33, 133.25, 132.62, 131.71, 130.03, 129.47, 128.72, 128.30, 128.24, 128.15, 127.49, 127.46, 127.36, 123.40, 122.81 (aromatic CH), 101.28 (C-1d), 83.90 (C-1c), 83.64 (C-3c), 79.85 (C-4c), 73.37 (PhCH2), 72.58 (C-2d), 71.04 (C-3d), 70.41 (C-5d), 69.87 (C-5c), 69.78 (C-6c), 65.33 (C-6d), 53.78 (C-2c), 32.25 (C-4d); FAB-HRMS (m/z) Calcd for C54H47NO13SNa (M + Na+): 972.2666. Found: 972.2687.
3.4 Phenyl (2,3,6-tri-O-benzoyl-4-deoxy-β-d-xylo-hexopyranosyl)-(1 → 4)-3-O-acetyl-6-O-benzyl-2-deoxy-2-phthalimido-1-thio-β-d-glucopyranoside (6a′)
A solution of 6a (100 mg) in 1 mL of pyridine and 0.5 mL of acetic anhydride was stirred at room temperature for 4 h and then concentrated, co-evaporated with toluene and dried. Compound 6a′ was obtained in quantitative yield (104 mg); Rf = 0.55 (toluene–ethyl acetate 4:1); [α]D +30 (c 0.7, chloroform); 1H NMR (400 MHz, CDCl3): δ 8.13–7.21 (m, 29H, aromatic), 5.78 (dd, 1H, J3,4 = 9.2 Hz, J2,3 = 10.1 Hz, H-3c), 5.67 (d, 1H, J1,2 = 10.6 Hz, H-1c), 5.39–5.35 (m, 1H, H-2d), 5.32–5.26 (m, 1H, H-3d), 4.78 (d, 1H, J1,2 = 7.9 Hz, H-1d), 4.71 (d, 1H, Jgem = 12.1 Hz, PhCH), 4.54–4.43 (m, 2H, H-6d, PhCH), 4.40–4.31 (m, 2H, H-2c, H-6d′), 4.11 (t, 1H, J3,4 = J4,5 = 9.2 Hz, H-4c), 3.90–3.84 (m, 1H, H-5d), 3.76 (dd, 1H, J5,6 = 3.3 Hz, Jgem = 11.2 Hz, H-6c), 3.64–3.57 (m, 2H, H-5c, H-6c′), 2.41 (ddd, 1H, J3,4 = 7.7 Hz, J4,5 = 1.9 Hz, Jgem = 12.6 Hz, H-4d), 1.90–1.80 (m, 4H, H-4d′, CH3); 13C NMR (100.6 MHz, CDCl3): δ 170.08 (CO, Ac), 167.73, 167.15, 166.19, 165.81, 164.88 (5CO, Bz, NPhth), 138.05, 131.69, 131.54, 131.23, 129.49, 129.27, 129.25 (aromatic C), 134.29, 134.07, 133.41, 133.23, 132.93, 129.69, 128.82, 128.65, 128.56, 128.42, 128.38, 128.04, 127.99, 127.83, 123.68, 123.50 (aromatic CH), 100.64 (C-1d), 83.05 (C-1c), 78.70 (C-5c), 75.32 (C-4c), 73.45 (PhCH), 72.49 (C-2d), 71.73 (C-3d), 71.65 (C-3c), 69.55 (C-5d), 67.51 (C-6c), 65.52 (C-6d), 53.98 (C-2c), 32.75 (C-4d), 20.46 (CH3, OAc); FAB-HRMS (m/z) Calcd for C56H49NO14SNa (M + Na+): 1014.2771. Found: 1014.2797.
3.5 Phenyl (2,3,6-tri-O-benzoyl-4-deoxy-β-d-xylo-hexopyranosyl)-(1 → 4)-[2,3,4-tri-O-benzyl-α-l-fucopyranosyl-(1 → 3)]-6-O-benzyl-2-deoxy-2-phthalimido-1-thio-β-d-glucopyranoside (8)
A mixture of 6a (1.19 g, 1.25 mmol, 1 equiv), 7 (1.52 g, 3.18 mmol, 2.5 equiv) and 4 Å powdered molecular sieves (2 g) in dry toluene (40.7 mL) was stirred at room temperature under argon for 40 min, then cooled to −20 °C. NIS (582.2 mg, 2.87 mmol, 2.3 equiv) and then TfOH (22.76 μL, 0.28 mmol, 0.23 equiv) were added. The mixture was stirred at −20 °C for 1 h, then neutralized with Et3N, filtered through Celite, and concentrated. The residue was washed with aqueous thiosulfate, water, brine and then dried over MgSO4 and concentrated. After purification by flash column chromatography (cyclohexane–ethyl acetate 5:1), compound 8 was obtained (0.97 g, 57%) as a white amorphous solid; Rf = 0.50 (cyclohexane–ethyl acetate 2:1); [α]D −1 (c 0.94, chloroform); 1H NMR (400 MHz, CDCl3): δ 8.14–7.09 (m, 44H, aromatic), 5.51 (d, 1H, J1,2 = 10.6 Hz, H-1c), 5.39–5.33 (m, 1H, H-2d), 5.30–5.24 (m, 1H, H-3d), 5.04 (d, 1H, J1,2 = 3.5 Hz, H-1e), 4.99 (d, 1H, J1,2 = 8.1 Hz, H-1d), 4.86 (d, 1H, Jgem = 12.0 Hz, PhCH), 4.80–4.76 (m, 2H, H-3c, PhCH), 4.70–4.66 (m, 1H, H-5e), 4.56–4.52 (m, 10H, H-2c, H-4c, H-6d, H-6d′, 2PhCH, 2PhCH2), 3.97–3.91 (m, 2H, H-3e, H-6c), 3.84–3.73 (m, 3H, H-2e, H-5d, H-6c′), 3.67 (d, 1H, J = 1.5 Hz, H-4e), 3.47 (d, 1H, J = 9.5 Hz, H-5c), 2.48–2.44 (m, 1H, H-4d), 1.77–1.69 (m, 1H, H-4d′), 1.33 (d, 3H, J5,6 = 6.5 Hz, H-6e); 13C NMR (100.6 MHz, CDCl3): δ 166.02, 165.77, 164.75 (3CO, Bz), 138.76, 138.23, 137.88, 132.30, 131.75, 129.60, 129.28, 129.24 (aromatic C), 133.98, 133.35, 133.21, 133.17, 132.34, 129.76, 129.68, 129.62, 128.70, 128.65, 128.55, 128.43, 128.35, 128.24, 128.21, 128.18, 128.05, 127.99, 127.97, 127.91, 127.88, 127.78, 126.83, 123.47 (aromatic CH), 99.50 (C-1d), 97.73 (C-1e), 83.94 (C-1c), 79.53 (C-3e), 79.13 (C-5c), 78.30 (C-4e), 75.14 (C-4c), 75.02 (C-2e), 74.75, 73.55, 72.79, 72.09 (4PhCH2), 74.69 (C-3c), 72.69 (C-2d), 71.67 (C-3d), 69.46 (C-5d), 67.86 (C-6c), 66.60 (C-5e), 65.54 (C-6d), 55.30 (C-2c), 33.32 (C-4d), 16.82 (C-6e); FAB-HRMS (m/z) Calcd for C81H75NO17SNa (M + Na+): 1388.4653. Found: 1388.4667.
3.6 2-(Trimethylsilyl)ethyl(2,3,6-tri-O-benzoyl-4-deoxy-β-d-xylo-hexopyranosyl)-(1 → 4)-[2,3,4-tri-O-benzyl-α-l-fucopyranosyl-(1 → 3)]-6-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranoside-(1 → 3)-(2,6-di-O-benzyl-β-d-galactopyranosyl)-(1 → 4)-2,3,6-tri-O-benzyl-β-d-glucopyranoside (10)
A mixture of 8 (50.7 mg, 0.037 mmol, 1 equiv), 9 (50 mg, 0.056 mmol, 1.5 equiv) and 4-Å powdered molecular sieves (100 mg) in dry dichloromethane (3.4 mL) was stirred at room temperature under argon for 40 min, then cooled to −30 °C. NIS (16.7 mg, 0.08 mmol, 2.2 equiv) and then TfOH (1.2 μL, 0.012 mmol, 0.33 equiv) were added at −30 °C. The temperature was raised slowly to room temperature during 30 min. The mixture was neutralized with Et3N after stirring at room temperature for an additional hour, filtered through Celite and then concentrated. The residue was washed with aqueous thiosulfate, water, and brine and then dried over MgSO4 and concentrated. After purification by flash column chromatography (toluene–ethyl acetate 9:1), compound 10 was obtained (74.7 mg, 94%) as a white foam; Rf = 0.64 (toluene–ethyl acetate 4:1); [α]D −1 (c 1.1, chloroform); 1H NMR (400 MHz, CDCl3): δ 8.12–7.20 (m, 64H, aromatic), 5.35 (dd, 1H, J1,2 = 8.0 Hz, J2,3 = 9.6 Hz, H-2d), 5.32 (d, 1H, J1,2 = 8.4 Hz, H-1c), 5.30–5.24 (m, 1H), 5.01 (d, 1H, J1,2 = 3.4 Hz, H-1e), 4.95 (d, 1H, J1,2 = 7.9 Hz, H-1d), 4.94–4.67 (m, 7H), 4.60 (dq, 1H, J4,5 < 1 Hz, J5,6 = 6.5 Hz, H-5e), 4.50–4.16 (m, 16H), 4.29 (d, 1H, J1,2 = 7.9 Hz, H-1a), 4.26 (d, 1H, J1,2 = 8.1 Hz, H-1b), 4.00 (sbr, 1H, H-4b), 3.98–3.81 (m, 5H), 3.76 (dd, 1H, Jgem = 10.1 Hz, PhCH), 3.68 (dd, 1H, J = 7.1 Hz, J = 9.5 Hz, 1H of OCH2), 3.63–3.56 (m, 2H), 3.52 (dd, 1H, Jgem = 10.5 Hz, PhCH), 3.49–3.29 (m, 9H), 3.02–3.00 (m, 1H), 2.73 (s, 1H, OH), 2.47–2.44 (m, 1H, H-4d), 1.76–1.67 (m, 1H, H-4d′), 1.29 (d, 3H, J5,6 = 6.5 Hz, H-6e), 1.03–0.98 (m, 2H, OCH2CH2Si), 0.02 (s, 9H, SiMe3); 13C NMR (100.6 MHz, CDCl3): δ 166.03, 165.76, 164.73 (3CO, Bz), 139.01, 138.74, 138.72, 138.48, 138.47, 138.26, 138.25, 137.59, 131.28, 129.23, 129.22 (aromatic C), 133.63–123.09 (64 aromatic CH), 102.95 (C-1b), 101.88 (C-1a), 99.48 (C-1d), 98.71 (C-1c), 97.61 (C-1e), 83.28, 82.79, 81.83, 79.44, 78.21, 77.96, 75.93, 75.35, 75.21, 74.64, 74.60, 73.70, 72.75, 72.51, 69.57 (CH), 75.29, 74.81, 74.73, 74.02, 73.74, 73.24, 72.84, 72.71, 72.07 (PhCH2), 71.63 (C-2d), 68.38, 67.94, 67.81, 67.13, 65.51 (4C-6, OCH2), 67.42 (C-4b), 66.62 (C-5e), 56.08 (C-2c), 33.25 (C-4d), 18.35 (OCH2CH2Si), 16.83 (C-6e), −1.49 (SiMe3); FAB-MS (m/z) Calcd for C127H133NO28SiNa (M + Na+): 2171.8714. Found: 2171.8.
3.7 2-(Trimethylsilyl)ethyl(2,3,6-tri-O-benzoyl-4-deoxy-β-d-xylo-hexopyranosyl)-(1 → 4)-[2,3,4-tri-O-benzyl-α-l-fucopyranosyl-(1 → 3)]-6-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranoside-(1 → 3)-(4-O-acetyl-2,6-di-O-benzyl-β-d-galactopyranosyl)-(1 → 4)-2,3,6-tri-O-benzyl-β-d-glucopyranoside (10′)
A solution of 10 (47.6 mg) in 1 mL of pyridine and 0.5 mL of acetic anhydride was stirred at room temperature for 12 h and then concentrated, co-evaporated with toluene and dried. Compound 10′ was obtained in quantitative yield (45 mg); Rf = 0.64 (toluene–ethyl acetate 4:1); [α]D −9 (c 1.2, chloroform); 1H NMR (400 MHz, CDCl3): δ 8.10–7.17 (m, 64H, aromatic), 5.42 (d, 1H, J = 3.5 Hz, H-4b), 5.38–5.32 (m, 1H, H-2d), 5.31–5.26 (m, 1H, H-3d), 5.23 (d, 1H, J1,2 = 8.3 Hz, H-1c), 5.07 (d, 1H, J1,2 = 8.0 Hz, H-1d), 5.00–4.94 (m, 1H), 4.95 (d, 1H, J1,2 = 3.4 Hz, H-1e), 4.90 (d, 1H, J = 2.2 Hz), 4.90–4.87 (m, 1H, H-4c), 4.77–4.66 (m, 5H, H-5e, H-3c), 4.58 (d, 1H, J = 12.1 Hz), 4.47–4.33 (m, 10H, H-2c), 4.31–4.21 (m, 7H, H-1a, H-1b), 4.04–3.95 (m, 2H, Jgem = 12.1 Hz, PhCH2), 3.89–3.73 (m, 5H, H-3e, H-5d, H-2e), 3.62 (sbr, 1H, H-4e), 3.57–3.51 (m, 1H), 3.50–3.30 (m, 10H), 3.01–2.98 (m, 1H, PhCH), 2.49–2.45 (m, 1H, H-4d), 2.09 (s, 3H, OAc), 1.79–1.71 (m, 1H, H-4d′), 1.31 (d, 3H, J5,6 = 6.4 Hz, H-6e), 1.04–0.99 (m, 2H, OCH2CH2Si), 0.03 (s, 9H, SiMe3); 13C NMR (100.6 MHz, CDCl3): δ 169.86 (CO, Ac), 166.01, 165.82, 164.83 (3CO, Bz), 139.08, 138.83, 138.73, 138.42, 138.26, 138.21, 131.35, 129.63, 129.52, 129.51 (aromatic C), 133.59–127.68 (64 aromatic CH), 103.02 (C-1b), 101.94 (C-1a), 99.50 (C-1d), 98.87 (C-1c), 97.35 (C-1e), 78.42 (C-4e), 75.09 (C-4c), 75.03 (C-5e), 73.11 (C-3c), 72.60 (C-2d), 71.81 (C-3d), 69.89 (C-4b), 69.45 (C-5d), 56.47 (C-2c), 82.60, 81.73, 79.48, 78.88, 78.71, 75.73, 74.59, 72.57, 66.48 (CH), 74.88, 74.76, 74.09, 73.66, 73.56, 73.44, 72.92, 72.60, 72.13 (PhCH2), 68.28, 67.68, 67.63, 67.21, 65.68 (4C-6, OCH2), 33.42 (C-4d), 20.76 (CH3, Ac), 18.39 (OCH2CH2Si), 16.83 (C-6e), −1.45 (SiMe3); FAB-MS (m/z) Calcd for C129H135NO29SiNa (M + Na+): 2213.8819. Found: 2213.8.
3.8 2-(Trimethylsilyl)ethyl(4-deoxy-β-d-xylo-hexopyranosyl)-(1 → 4)-[2,3,4-tri-O-benzyl-α-l-fucopyranosyl-(1 → 3)]-6-O-benzyl-2-deoxy-2-acetamido-β-d-glucopyranoside-(1 → 3)-(2,6-di-O-benzyl-β-d-galactopyranosyl)-(1 → 4)-2,3,6-tri-O-benzyl-β-d-glucopyranoside (11)
Hydrazine monohydrate (1.5 mL) and water (1.5 mL) were added to a solution of compound 10 (140 mg, 0.065 mmol) in 10 mL of ethanol. The mixture was refluxed for 14 h. After concentration, the residue was co-evaporated with toluene and dried, then dissolved in 4 mL of methanol–dichloromethane (1:1), to which 0.5 mL of acetic anhydride was introduced. The mixture was stirred at room temperature for 2.5 h. After concentration, the residue was purified by a column of silica gel, eluted with ethyl acetate and then by a Sephadex column (LH20) using methanol–dichloromethane (1:1) as eluant. Compound 11 was obtained (101 mg, 89%) as a white amorphous solid; Rf = 0.24 (ethyl acetate); [α]D −19 (c 1.1, chloroform); 1H NMR (400 MHz, CDCl3): δ 8.10–7.17 (m, 45H, aromatic), 5.27 (d, 1H, J1,2 = 7.3 Hz, H-1c), 5.10–5.02 (m, 2H, H-1e, PhCH), 5.00–4.92 (m, 3H, H-3e), 4.83–4.78 (m, 4H, H-2e, 3PhCH), 4.74–4.57 (m, 8H, H-2a), 4.51–4.45 (m, 2H, H-1d, H-1a), 4.41 (d, 1H, J1,2 = 9.1 Hz, H-1b), 4.51–4.33 (m, 5H), 4.12–3.93 (m, 7H), 3.91–3.88 (m, 1H, H-5c), 3.76–3.54 (m, 17H), 3.49–3.35 (m, 4H), 3.22–3.14 (m, 2H), 1.85 (dd, 1H, Jgem = 12.3 Hz, J3,4 = 4.9 Hz, H-4d), 1.41 (s, 3H, OAc), 1.41–1.33 (m, 1H, H-4′d), 1.18 (d, 3H, J5,6 = 6.4 Hz, H-6e), 1.12–1.07 (m, 2H, OCH2CH2Si), 0.04 (S, 9H, SiMe3); 13C NMR (100.6 MHz, CDCl3): δ 170.74 (CO, Ac), 139.08, 138.84, 138.70, 138.42, 138.30, 138.31, 138.27, 138.03, 137.27 (aromatic C), 128.49–127.07 (aromatic CH), 102.99 (C-1b), 102.10 (C-1a), 99.62 (C-1d), 99.50 (C-1c), 97.99 (C-1e), 82.83, 82.18, 81.86, 79.27, 78.89, 77.19, 76.62, 76.40, 76.23, 75.85, 75.74, 74.96, 74.73, 73.54, 72.83, 70.86, 67.81, 67.36 (ring CH), 75.21, 74.84, 74.55, 73.92, 73.51, 73.28, 73.04, 72.48 (PhCH2), 70.06, 68.77, 68.25, 67.21, 64.99 (4C-6, OCH2CH2), 57.26 (C-2c), 33.22 (C-4d), 22.78 (CH3, Ac), 18.38 (OCH2CH2Si), 16.66 (C-6e), −1.47 (SiMe3); FAB-HRMS Calcd for C100H121NO24SiNa (M + Na+): 1770.7946. Found: 1770.7969.
3.9 2-(Trimethylsilyl)ethyl(4-deoxy-β-d-xylo-hexopyranosyl)-(1 → 4)-[α-l-fucopyranosyl-(1 → 3)]-2-deoxy-2-acetamido-β-d-glucopyranoside-(1 → 3)-(β-d-galactopyranosyl)-(1 → 4)-β-d-glucopyranoside (1)
A solution of 11 (40 mg) in methanol (3.85 mL) was treated with Pd/C (10%, 13.5 mg) under H2 (160 kPa) for 13 h at 25 °C, then filtered and evaporated. The residue was purified on a Sephadex column (G25) using water as eluant. Compound 1 was obtained (20.4 mg, 95%) as a white amorphous solid; Rf = 0.25 (isopropanol–ethyl acetate–water 3:3:1); [α]D −65 (c 0.88, chloroform–methanol 1:1); 1H NMR (400 MHz, D2O): δ 5.13 (d, 1H, J1,2 = 4.0 Hz, H-1e), 4.86–4.80 (m, 1H, H-5e), 4.71 (d, 1H, J1,2 = 8.3 Hz, H-1), 4.50 (d, 1H, J1,2 = 8.0 Hz, H-1), 4.46 (d, 1H, J1,2 = 7.8 Hz, H-1), 4.44 (d, 1H, J1,2 = 7.8 Hz, H-1), 4.16 (d, 1H, J = 3.3 Hz), 4.07–3.53 (m, 25H), 3.31–3.27 (m, 1H, H-2), 3.11 (dd, 1H, J1,2 = 8.0 Hz, J2,3 = 9.0 Hz, H-2), 2.02 (s, 3H, Ac), 1.99 (dd, 1H, Jgem = 11.3 Hz, J3,4 = 5.1 Hz, H-4d), 1.29–1.21 (m, 1H, H-4d′), 1.20 (d, 3H, J5,6 = 6.6 Hz, H-6e), 1.03 (2dt, 2H, Jgem = 12.9 Hz, Jvic = 5.5 Hz, CH2Si), 0.034 (s, 9H, SiMe3); 13C NMR (100.6 MHz, D2O): δ 175.10 (CO, NHAc), 103.30, 102.91, 102.04, 101.73 (C-1a, C-1b, C-1c, C-1d), 99.14 (C-1e), 82.45, 78.65, 75.49, 75.45, 75.23, 75.11, 74.89, 73.51, 72.73, 72.34, 70.48, 70.33, 69.59, 68.67, 68.06, 66.81 (ring CH), 75.81 (C-2), 73.18 (C-2), 68.81 (OCH2CH2Si), 64.53, 61.32, 60.41, 59.97 (C-6a, C-6b, C-6c, C-6d), 56.34 (C-2c), 35.07 (C-4d), 22.63 (CH3, Ac), 17.93 (CH2Si), 15.80 (C-6e), −2.16 (SiMe3); FAB-HRMS (m/z) Calcd for C37H67NO24SiNa (M + Na+): 960.3720. Found: 960.3708.
Acknowledgements
We thank the French Embassy in China for a Ph.D. fellowship to Y. Luo, the China Scholarship Council and Guizhou University for a Ph.D. fellowship to D. Dong. Financial support from CNRS, ENS and ZJU is gratefully acknowledged.