

1 Introduction
As natural polyfunctional and chiral molecules, carbohydrates have been the subjects of intensive researches for quite a long time. They are continuously attracting much attention from synthetic chemists, in particular for designing shorter stereocontrolled routes towards oligosaccharides and other molecules of interest for glycosciences and medicine [1]. Due to the presence of various nucleophilic groups (e.g., hydroxyl, amine) in sugars, protection/deprotection strategies are generally required to prevent unwanted reactions. The search of milder and orthogonal conditions has inspired most advances, in particular for stereocontrolled glycosidations [2]. Ionic approaches were first more popular and many methods have been proposed for the generation of d-glycopyranosylium ions as reactive intermediates for glycosylation reactions, to afford O-, N-, or S-glycosides, among other structures. C-Glycosyl compounds (also frequently termed C-glycosides) display an anomeric carbon constitutive of an ether motif, and are, compared to common glycosides, more stable to both acid and enzyme-catalyzed hydrolysis, because of the resistance of ethers compared to acetals [3]. C-Glycosyl compounds, and among them C-glycosyl flavonoids are well represented in Nature, some of them having interesting bio-activities [4]. Therefore, their synthesis is well documented, based on a large variety of versatile approaches. Electrophilic substitution offers, in particular with electron-rich aryls, a suitable route towards C-glycosyl arenes with possible stereocontrol [5].
After 1980 or so [6], the synthetic potential of free-radical methods became more appreciated, thus inspiring numerous investigations of sugar-based radicals, as regard to their structure, their synthetic transformations under very mild conditions, and their participation in biological events. Our interest in radical-based transformations of sugar derivatives stems from early investigations carried out in the group of Descotes, who first reported efficient conversions of alkyl glycosides into sugar-based spiroacetals or spiro-orthoesters via, respectively, carbon, or oxygen-centered radical intermediates generated with UV or visible-light irradiation [7]. From these studies of intramolecular reactions generally based on d-gluco configured precursors, it was concluded that the newly created bond was established with high α-stereoselectivity. Intermolecular processes also occurred with high α-stereoselective bond formation [6], as the tri-n-butyltin deuteride reduction of glycosyl halides [8] or the NBS-mediated bromination of various glycosyl derivatives, as β-d-glycopyranosyl cyanides [9] or chlorides [10]. It is worth mentioning that protection of the hydroxy groups by acetylation or benzoylation proved to be suitable under the conditions promoting the aforementioned radical reactions, so that NBS-mediated bromination of 2,3,4,6-tetra-O-acetyl-β-d-glycopyranosyl chlorides afforded the corresponding 2,3,4,6-tetra-O-acetyl-1-bromo-β-d-glycopyranosyl chlorides in ∼65% yield [10]. Such sugar dihalides have been converted to sugar-based orthoesters [11], alkoxyazides [12], diazides [13], which allowed unprecedented ring expansions [14] or ring formation [15]. Sugar dihalides also proved to be synthetically useful for the stereocontrolled synthesis of C-β-d-glycosides, as described below.
This account summarizes our recent achievements in the field of C-glycosyl compounds, made possible with the participation, for the synthetic work, of co-tutored students from the group of Prof. Guo-Rong Chen, East China University of Science and Technology, Shanghai, PR China, as presented at 1st Chemical Workshop South China–Lyon. Other established collaborations allowed appropriate biological evaluations.
2 Radical synthesis of C-glycopyranosyl-alkyl compounds
In addition to ionic approaches [3], radical-based syntheses of C-glycopyranosyl-alkyl compounds have received considerable attention and many examples have been reported, thus highlighting the potential of radical methods [3,6]. These reactions generally involve reagents of the organotin type (e.g., allyl tri-n-butyltin, tri-n-butyltin hydride) transformed in the reaction into organotin products, which are toxic and difficult to remove at the purification stage. Therefore, the development of mild methods using either no organotin compounds or catalytic amounts, but affording the products with good (stereo)selectivity was worth of consideration.
2.1 Synthesis of alkyl C-α-d-glycosides by radical addition to alkenes
While carbon-centered glycosyl radicals add smoothly to allyl tri-n-butyltin in a chain reaction based on fragmentation of radical intermediates that produce the chain-carrying tri-n-butyltin radical [6] and C-glycosyl-propenes [16], the possibility for glycosyl radicals to add to other alkenes offers extended applications. Due to the ring oxygen next to the radical center, glycosyl radicals are nucleophilic and react easily with electron-poor alkenes e.g. acrylonitrile, acrylic esters, diethyl vinylphosphonate. The radical intermediates thus formed by addition, with (OR having) the radical center located next to an electron-withdrawing group (e.g. cyano, ester, phosphonate) are electron-poor and, being electrophilic, they react fast with tri-n-butyltin hydride. This step delivers the reaction product and a new tin centered-radical to continue the chain reaction. A fast addition of the glycosyl radical to the alkene is crucial for the success of C–C bond forming reaction, which can otherwise suffer from radical reduction, among other competing reactions, in particular with 2-deoxy-2-amino sugars [17]. Reports on the synthesis of C-glycosyl compounds by addition of glycosyl radicals to alkenes showed some limitations, as the moderate thermal stability of glycosyl halides used as the radical precursors and the possible influence of the solvent [18]. In the quest of optimized conditions, UV-light initiation of the reaction appeared advantageous, as well as use of tri-n-butyltin hydride in catalytic amounts, with NaBH3CN in excess. NaBH3CN can reduce the tin halide (chloride, bromide) formed during the reaction so as to maintain a low concentration of tin hydride as required to continue the radical chain reaction. t-Butanol is the solvent of choice, due to its low absorbance at the wavelengths used and as it dissolves NaBH3CN [19].
Our observations [20] clearly show (Scheme 1) that under optimized conditions at mild temperatures (30/35 °C) the addition of d-glycopyranosyl radicals to acrylonitrile [18,21] and diethyl vinylphosphonate [22] afforded efficiently the corresponding α-configured C-glycosyl compounds and only trace amounts of the β-anomers. With both alkenes, the recorded yields were very similar, and somehow higher than those previously reported, either with acrylonitrile (57–76%) [21], or diethyl vinylphosphonate (<44% yields in Ref. [22a]). Among the minor products isolated, we obtained both 1,5-anhydro-alditol 4 and 2-deoxy sugar 5 which resulted from the reduction of either the initial anomeric radical or that formed by 2,1-acetoxy migration [8c]. To our delight, in the absence of acrylonitrile, these reductions predominated and could be controlled tightly (Scheme 2) under simple conditions without any tin compound added: photoirradiation of 1 in t-BuOH in the presence of NaBH3CN led in 82% yield to a 95/5 5/4 crude mixture, while adding catalytic amounts of thiophenol prevented the radical rearrangement because of polarity reversal catalysis [8d], to afford a 98/2 mixture of 4/5 in 79% yield [20].
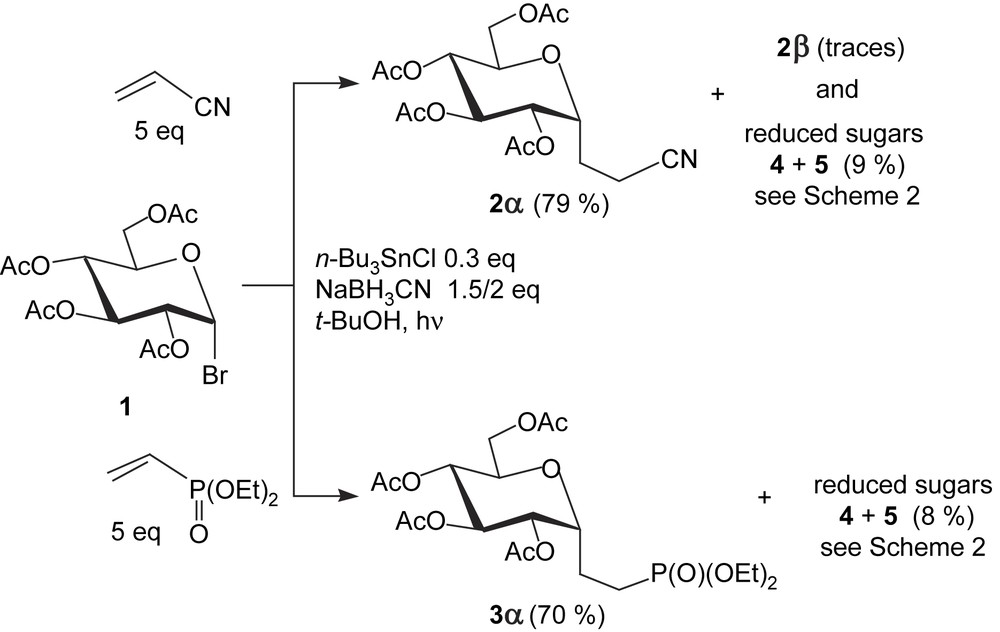

(i) NaBH3CN 2 equiv, t-BuOH, hν, 0.5 h; (ii) NaBH3CN 2 equiv, C6H5SH 10 mol%, t-BuOH, hν, 1.75 h.
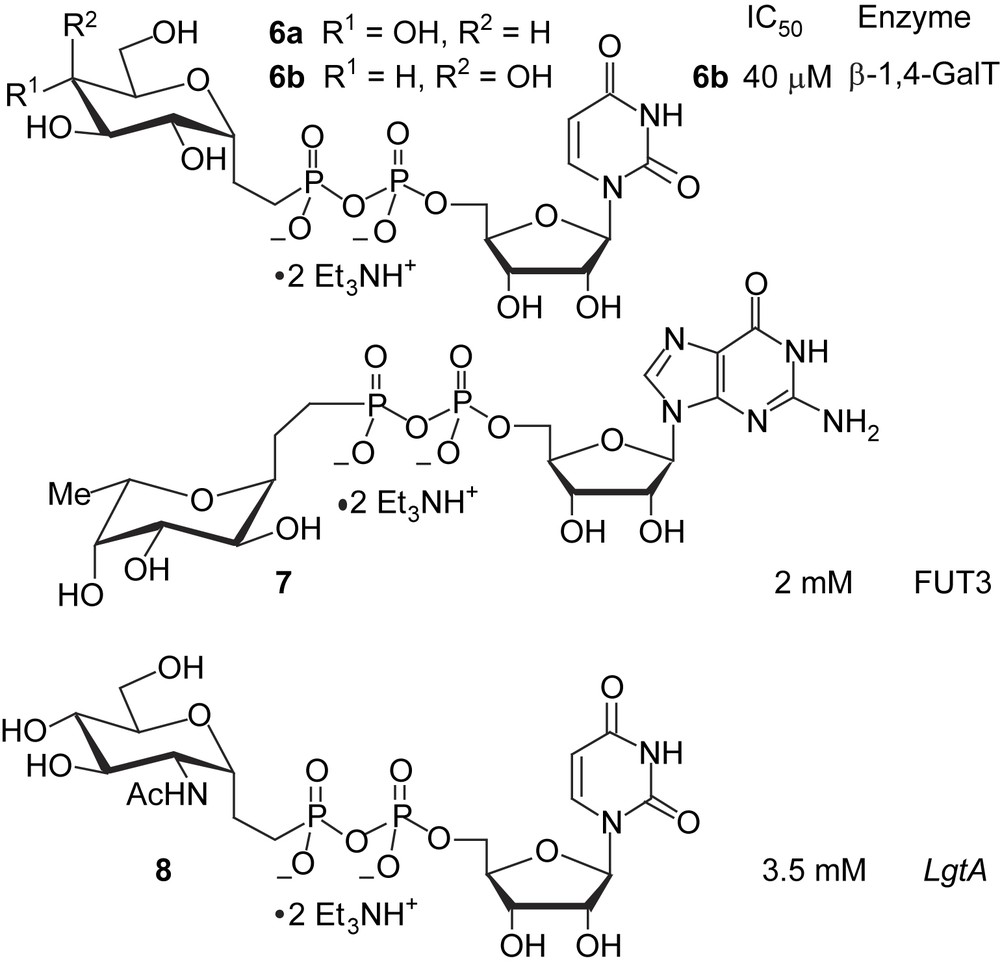
The accessibility of such C-glycosyl-ethylphosphonates made possible the synthesis of C-glycosyl analogues (Scheme 3) of natural substrates of glycosyl transferases. Coupling of deprotected glycosyl ethylphosphonic acids with activated nucleotides (UMP-morpholidate or GMP-morpholidate) was the key step in the sequence, achieved with 10/24% yields [23]. Enzymatic evaluation with suitable glycosyl transferases [24] showed that 6b was a good inhibitor (IC50 = 40 μM) of bovine milk β-1,4-galactosyltransferase (β-1,4-GalT, EC 2.4.1.22).
2.2 Stereocontrolled synthesis of C-β-d-glycosides
The aforementioned conditions applied to prepare C-α-d-glycosyl compounds appeared favorable to extend further the scope of radical methods. In particular, the available peracetylated sugar dihalides [10] appeared valuable substrates for the stereocontrolled synthesis of C-β-d-glycosyl compounds. We supposed that upon reaction of sugar dihalides (e.g. 9) with tin radicals, homolysis of the C–Br axial bond should be easier as compared to the equatorial C–Cl bond (anomeric effect, BDE lower for the C–Br bond), thus leading to chlorinated anomeric radicals. Their addition to either allyl tributyltin or acrylonitrile/diethyl vinylphosphonate (in the presence of n-Bu3SnH to reduce the so-formed radical intermediate) would lead to chlorinated C-glycosyl compounds. Although prone to hydrolysis, they could be obtained, sometimes in high yield, as for 10, apparently more stable than its d-galacto/d-manno analogues (51% and 31% isolated yield, respectively). However, radical reduction of the chlorinated C-glycosyl compounds with an excess of n-Bu3SnH (one-pot or stepwise process) occurred as expected with an α-stereoselectivity, as shown by the β-anomeric configuration of the obtained products 2β, 3β, 11 (Scheme 4). In this way, besides minor products formed by hydrolysis, radical rearrangement, and reduction, the desired β-configured anomers were obtained as the major products (38/55%) in comparable yields, independently of the sugar configuration [20,25].

(i) Allytributyltin 2 equiv, C6H6, hν; (ii) n-Bu3SnH 2 equiv, C6H6, hν; (iii) CH2CHCN or CH2CHP(O)(OEt)2 5 equiv, n-Bu3SnCl 0.3 equiv, NaBH3CN or Bu4NBH3CN 2 equiv, t-BuOH or C6H6, hν; (iv) C6H6, n-Bu3SnH, 3/4 equiv, hν, 15 min.
2.3 Synthesis of glycodienes, bis-C,C-glycosides and spiro derivatives
Interestingly, acetylated 1-chloro-C-glycosyl propenes 10, 16, 17 (d-gluco, d-galacto, d-manno configurations) underwent DBU-induced elimination of hydrochloric acid, to form hitherto unprecedented (Z)-configured glycodienes [25,26]. Since C-ketosyl compounds and bis-C,C-glycosides are not very common structures, with limited accessibility [27], sugar dihalides appeared candidates of choice for preparing under very mild conditions bis-C,C-glycosides having two allyl residues (Scheme 5). Applying to 9 and related sugar dihalides the C–C bond forming conditions with excess of allyltributyltin led in moderate yield to the desired bis-C,C-glycosides, which were converted efficiently by the ring-closing metathesis reaction with Grubb's catalyst to d-gluco, d-galacto, and d-manno glycosyl-spiro-cyclopentenes [28]. The new structures prepared were deacetylated readily.

3 C-Glycosyl-hydro(benzo)quinones and derivatives
Because of their natural occurrence and due to the existence of several synthetic approaches to C-glycosyl arenes, such compounds are well documented, in particular as phenol or polyphenol (resorcinol, phloroglucinol) derivatives [5]. Within a project focusing on C-glycosyl compounds with potential antithrombotic activities [29], we noticed that C-glycosyl derivatives derived from hydro(benzo)quinones are poorly known, except for the early investigation reported by Kalvoda [30], who mainly described furanosides of hydroquinone and C-ribofuranosyl derivatives of dimethoxybenzenes and hydro(benzo)quinone. While the O→C rearrangement of hydroquinone glycosides to C-glycosides appears to be unprecedented, scattered reports described the synthesis of C-glycosyl derivatives of hydroquinone and analogues based on carbene or radical intermediates. Considering that C-glycosyl aryls are stable molecules prone to various chemical modifications that may lead to bioactive structures [31], we decided to focus our efforts on the synthesis of C-glycopyranosyl aryls by electrophilic substitution of 1,4-dimethoxybenzene [32]. The so-obtained molecules could be converted into a variety of stable C-glycosyl derivatives interesting for both chemical modifications and bioactivities. Initially, we planned to synthesize glucose-based molecules for biological evaluation as glycogen phosphorylase (GP) inhibitors [33] for study in the frame of an established collaboration [34].
3.1 Synthesis of C-glycosyl-hydro(benzo)quinones
As mentioned before, C-glycosyl-hydro(benzo)quinones are simple structures which are poorly known, except for one report by Kalvoda, who studied a route based on electrophilic substitution of dimethoxybenzene by glycofuranosyl cations followed by oxidation and reduction [30]. Electrophilic substitution of 1,4-dimethoxybenzene with glycopyranosylium ions [32] followed by oxidation and reduction was therefore investigated [35]. The coupling reaction of glycopyranosyl cations and 1,4-dimethoxybenzene can be promoted by SnCl4 and various salts, in particular F3CCOOAg in CH2Cl2 as solvent [32]. In order to optimize, in terms of yield and β-stereoselectivity, such a C-glycosylation applied to 27 and 28, we found that the reaction temperature was critical. So as to shift equilibria towards the thermodynamically favoured β-anomers within a reasonable time, mild heating to 25/30 °C was applied for 4/5 h, resulting in reproducible yield of the desired compounds (d-gluco: 69%; d-galacto: 81%). Oxidation with ceric ammonium nitrate (CAN) afforded cleanly the corresponding glycosyl-benzoquinones (Scheme 6) whose deacetylation proved to be unselective, except for the unexpected formation of chlorinated hydroquinone derivatives on stirring in MeOH acidified with AcCl. Therefore, in view of preparing deprotected glycopyranosyl-benzoquinones 37, 38 for biological evaluations, the oxidation, reduction and deacetylation steps were combined in different sequences: (a) the dimethoxy derivatives 29β and 30β were deacetylated, then oxidized (CAN); (b) the benzoquinones 31, 32 were reduced, deacetylated, then oxidized, either by Ag2O or by PhI(OAc)2. This afforded deacetylated glycopyranosyl benzoquinones which were tested without delay, because of their limited stability.

(a) 5 or 6, p-Dimethoxybenzene 2 equiv, SnCl4 3 equiv, CF3CO2Ag 1.5 equiv, CH2Cl2, 20–25 °C, ∼ 5 h; (b) CAN 3 equiv, CH3CN/H2O, 1:1 or 2:3, 25 min; (c) MeOH containing 0.5% AcCl, 1 week, rt; (d) MeOH/NEt3/H2O, 8:1:1, rt, 10/12 h; (e) for 29β/30β or 39/40, respectively, 0.1 M or 0.033 M MeONa in MeOH, rt, ∼ 2 h; (f) CAN 3 equiv, H2O, rt, 30 min; (g) NaBH4 2 equiv, EtOAc, rt, 30 min; (h) MeOH containing 1% AcCl, 5 days, rt; (i) Ag2O (8 equiv with 41, oxidation almost complete; 3 equiv with 42, ∼95% conversion), 2-propanol, rt, 2 h; (j) PhI(OAc)2 1.5 equiv, MeOH, rt, 40 min; (nd) not determined.
3.2 Synthesis of C-glycosyl-chromanols and C-glycosyl-tocopherols
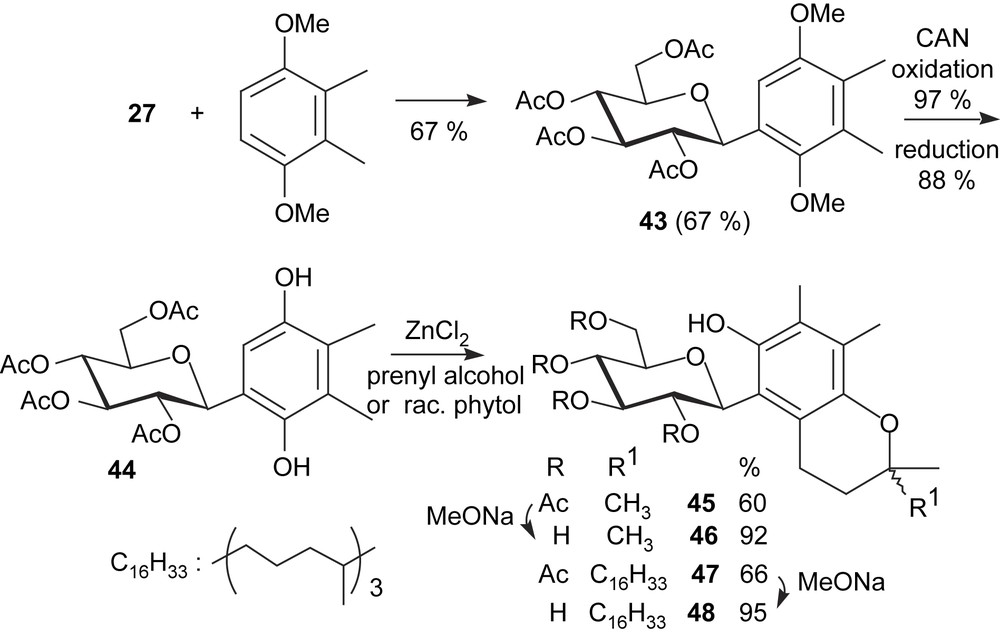
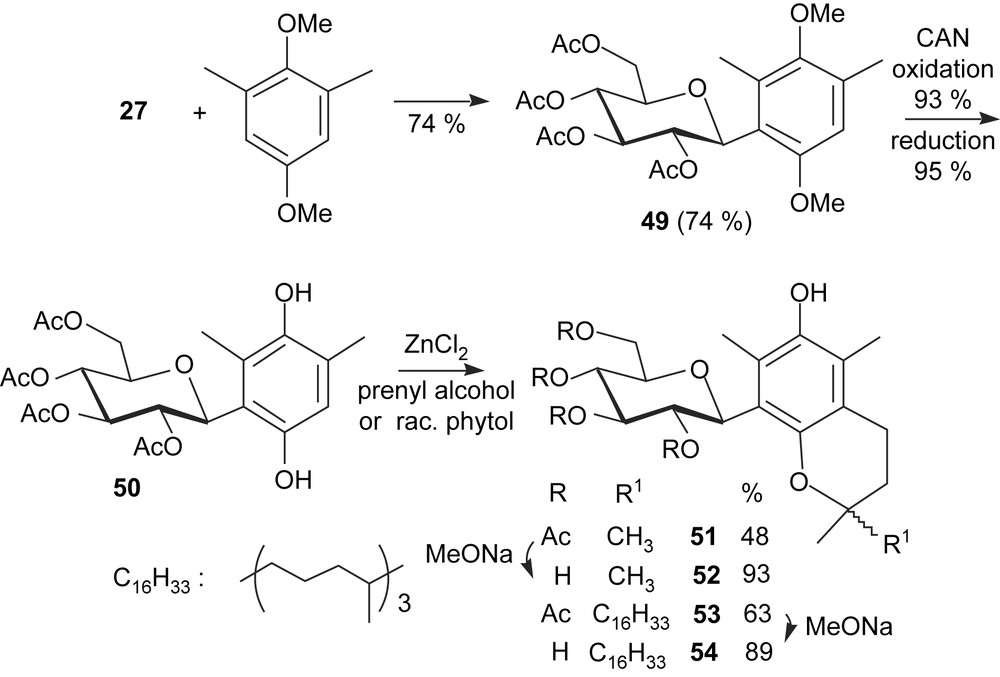
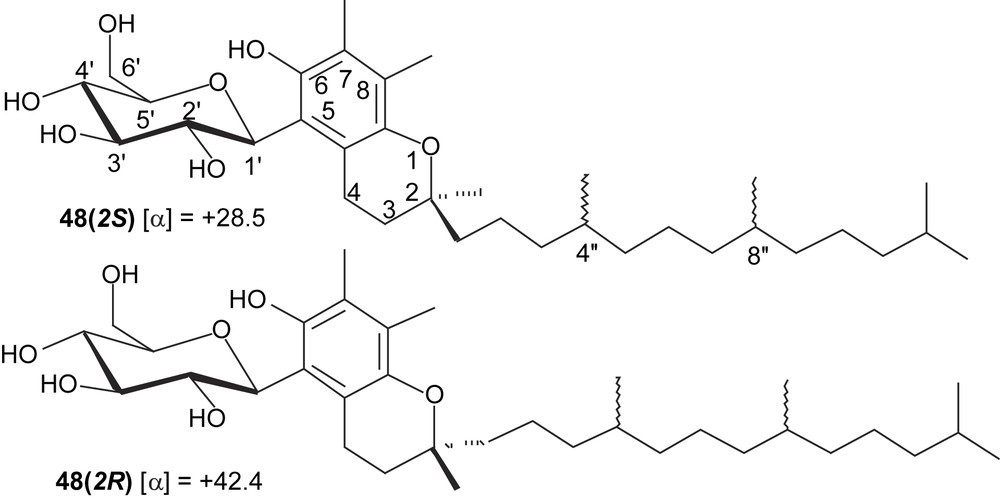
Among free-radical scavengers and antioxidants, vitamin E is well known, particularly because α-tocopherol is the more abundant and the more effective dietary antioxidant, with some industrial importance. Its structure makes it essentially lipophilic. Modified analogues with polar residues have been prepared, and shown to dissolve in polar solvents, sometimes in water, as it is the case of tocopheryl oligosaccharides. However, such O-glycosides have no free phenoxy group and they are dependent on in vivo deglycosidation to exert antioxidant properties. This problem does not exist in the case of C-glycosyl-tocopherols. To test their synthesis, hydroquinone 39 was reacted with racemic phytol in the presence of ZnCl2. Because both phenoxy groups could participate in cyclization, this attempt afforded a mixture of glycosylated compounds, which proved to be difficult to resolve by chromatography. To avoid such unselective annelations, defined glycosylated dimethylhydroquinones were prepared, and syntheses were adapted to isomeric o-, m-, and p-dimethyl-1,4-dimethoxybenzenes (Schemes 7 and 8). The first one was prepared from commercially available o-dimethylhydroquinone by methylation, while high-yielding three-step sequences (oxidation with Fremy's salt, reduction, methylation) applied to 2,6-, or 2,5-dimethylanisole led, respectively, to the other precursors. While the electrophilic substitution of o-, and m-dimethyl-1,4-dimethoxybenzenes with glucopyranosyl cation was effective (67 and 74% yield, respectively), use of the para isomer afforded an α/β anomeric mixture of C-glucosyl compounds in <17% yield, thus hampering the next steps. The rest of the sequence [CAN oxidation, reduction, annelation with 3-methyl-2-buten-1-ol (prenyl alcohol) or racemic phytol] applied to 44 and 50 was straightforward, as were the final deacetylations under Zemplén conditions, so that various C-glucopyranosyl chromanol derivatives were obtained. With racemic phytol, these sequences afforded mixtures of diastereoisomers, with an R or S configuration at C2, C′′4, and C′′8 (Scheme 9), as evidenced by examination of high-field NMR spectra. In the case of deacetylated compound 48, column chromatography allowed the separation of the 2R and 2S diastereoisomers (Scheme 9).
3.3 Biological evaluation: preliminary tests
The prepared molecules were subjected to different tests. C-Glycopyranosyl-hydro(benzo)quinones were assayed as protein tyrosine phosphate 1B (PTP1B) inhibitors [36], the main reason being that several quinones are known to inhibit PTP1B via a specific mechanism [36b]. Being responsible for the dephosphorylation of the insulin receptor, a process which turns off the insulin signaling cascade, PTP1B is considered as a promising target in therapeutic approaches towards non-insulino-dependent diabetes (type-2 diabetes) [37]. Deacetylated d-glucopyranosyl-hydro(benzo)quinones were also tested as glycogen phosphorylase (GP) inhibitors. Since GP catalyzes glycogen depolymerization, it was assumed that glucose-based molecules could fit the enzyme active site, while the aglycon could establish stabilizing non-covalent interactions at its vicinity, with the result of competitive inhibition [33,34]. Finally, the glycosyl-chromanols and glycosyl-tocopherols were investigated with the expectation that their structural features (type of substitution, free or acetylated sugar ring, Me versus the C16H33 chain) will modulate their expected antioxidant properties.
3.3.1 Inhibition of PTP1B
C-Glycopyranosyl-benzoquinones proved to be irreversible inhibitors of PTP1B in vitro, as found by Prof. Jia Li, Chinese National Center for Drug Screening, Shanghai Institute of Materia Medica, Shanghai, PR China [38]. The acetylated β-d-glucosyl-benzoquinone 31 was the best inhibitor of PTP1B (IC50 = 4.8 μM), as compared to the deacetylated d-gluco and d-galacto analogues 37 and 38 (IC50 = 25.6 and 24.3 μM, respectively). Unfortunately, other analogues such as β-d-glucopyranosyl-naphthoquinones showed no activity in these tests. Ongoing efforts are pursued to explore in more detail such an interesting inhibition, having in mind, however, that potent molecules or leads suitable for pharmaceutical development should be bioavailable intracellularly, with a high selectivity to PTP1B compared to other phosphatases, among other criteria [36].
3.3.2 Inhibition of glycogen phosphorylase
Enzymatic evaluations of 35, 37, and 39 against GP showed modest inhibitions for 37 and 39 with Ki = 3.8 and 2.6 mM, respectively, while 35 had no activity. However, after soaking the crystals of GP in the presence of 37 and 39 as ligands, it was possible to achieve the crystal structure analysis of the obtained ligand–enzyme complexes. The β-d-glucosyl-hydro(benzo)quinones were found to bind at the enzyme active site [35b]. Even though the measured inhibitions were modest, such molecules represent a new type of GP inhibitors with specific features associated to the C-glycosyl arene linkage (stability, conformers with restricted rotation). In the quest of more active molecules, our ongoing efforts focus on synthetic modifications of the aglycon moiety.
3.3.3 C-glycosyl-tocopherols as antioxidants
As noted before, the naturally occurring tocopherols and tocotrienols (vitamin E), with a phenoxy group and C16 alkyl chains (saturated or not), are major lipophilic antioxidants, among other species from the diet, which act in connection with the hydrophilic ascorbic acid (vitamin C) [39]. Because of the biological importance of free radicals, as regard to normal processes, damages to biomolecules and tissues or aging, this field is the focus of intensive researches. While a better insight into the phenomena is still necessary [40], the idea that free radicals contribute to health has reached a consensus (French paradox, beneficial effects of tea). As a consequence, the food and health companies are strongly involved in basic research and in the development of improved products, which may incorporate supplements, as nutraceutics.
The obtained glycosylated chromanol and tocopherol analogues were evaluated as antioxidants by Galland [41] at the University of Avignon (France) under the supervision of Dangles [42]. The observations collected in the case of the linoleic acid peroxidation point to antioxidant activities lower than that of tocopherol, as shown by the following the sequence: α-tocopherol > 51 ≈ 54 > 53 > 52 > 45 > 47 > 48 > 46. The synthetic data and a detailed analysis of the antioxidant properties will be reported in a forthcoming article, in preparation [43].
Acknowledgements
On the occasion of the 20th anniversary of the scientific and cultural collaboration between "Région Rhône-Alpes" (France) and the Shanghai City Council (PR China), generous financial support, and in particular stipends ["Mobilité internationale Rhône-Alpes" studentships to Fei ZhongBo, Qin Bing Bing (1999), He Li, Zhang Yun Zhi (2002–2005), Xue JiaLu (2006)] are gratefully acknowledged. Supports from the University Claude-Bernard–Lyon-1 for co-tutored thesis, from the French Ministry of Foreign Affairs (ARCUS Chine 2005), and from the National Science Foundation of China (Grant No. 20576034) are also acknowledged.