

1 Introduction
In the search for rapid conversion of carbohydrates into useful synthons, our group has been interested in the chemistry of palatinose, a disaccharide which is cheap, readily available and produced by bioconversion on an industrial scale from sucrose. In particular, we have shown that the hydrogen peroxide oxidation of isomaltulose offers a convenient access to carboxymethyl α-d-glucopyranoside (α-CMG) [1], an interesting synthon for coupling a carbohydrate moiety onto various chemical architectures [2–7]. Also, we found that the treatment of α-CMG with acetic anhydride provided the corresponding 3,4,6-tri-O-acetyl-α-d-glucopyranoside 2-O-lactone (α-CMGL), which has been shown to serve as an interesting electrophilic species for the synthesis of pseudoglycoconjugates. For example, opening of the lactone ring of CMGL by various amines allowed one to prepare glycocaminoacid hybrids [8], pseudodissacharides [9] and pseudoglucolipids [10]. Besides the simplicity of the reaction, a simple addition which does not require any activating reagent, the glycoconjugate-amides display a free OH function at the 2 position of the glucose moiety, which can be used for further substitution.
The synthetic interest of such bicyclic lactones has led us to work in two directions, one dealing with the preparation of analogous compounds based on other carbohydrate backbones, and the other, being described here, dealing with the use of either isomaltulose or its hydrogenated analogue, isomalt®, as starting materials, the possible side products observed when the oxidation step is either incomplete or excessive, the formation of the lactone and the by-products observed depending on the conditions, the stability of the lactone, and finally the synthesis of lactones having different ester protecting groups to the acetyl ones.
2 Results and discussion
Previous studies on palatinose (1) and trehalulose [1] have shown that oxidation gave the best results in acidic conditions using hydrogen peroxide and catalytic amounts of sodium tungstate following a modified version of the Venturello–Ricci–Noyori method [11]. Reaction was performed at 80 °C for 25 h and in the case of palatinose, even if the α-inter-glycosidic linkage is not very prone to cleavage under acidic conditions, the reaction mixture was still maintained at pH 4 to give fair yields of α-CMG (2). The reaction was followed by HPLC and the maximum yield observed was 38% [1]. When the mixture obtained containing the intermediate 2 was acetylated using pyridine and acetic anhydride, compound 3 was obtained in 29% overall yield from 1 (Scheme 1).
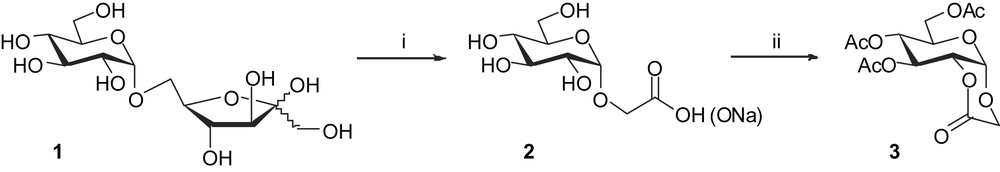
Reagents and conditions: (i) H2O2, W2O4, H3PO4, 90 °C, 15 h; (ii) Ac2O, pyridine, r.t., 15 h.
Our initial goal for this study was directed towards a better understanding of the oxidation step of this two-step transformation by analysis of the side products formed. Nevertheless, compounds were identified after acetylation and a selection of the results is summarized in Table 1.
Selected results obtained using different reaction conditions
| Entries/Starting compounds | Oxidation | Acetylation | Obtained compounds | |||||||
| pH | T (°C) | NaWO4 | Reaction times (h) | Quenching reagents | Solvents | Reaction times (h) | 3 (%) | 4 (%) | 5 (%) | |
| 1/1 | 4 | 90 | Yes | 16 | Na2SO3 | Pyridine | 48 | 23 | + | n.d. |
| 2/1 | 2 | 80 | No | 14 | Na2SO3 | Pyridine | 72 | 12 | 8 | 3 |
| 3/1 | 2 | 80 | No | 22 | MnO2 | Pyridine | 12 | 8 | 10 | n.d |
| 4/1 | 4 | 90 | Yes | 48 | MnO2 | DMF/Et3N | 15 | 23 | + | n.d. |
| 5/11 | 4 | 90 | Yes | 100 | MnO2 | Pyridine | 48 | 12 | 2 | + |
| 6/11 | 2 | 90 | Yes | 20 | MnO2 | Pyridine | 48 | 16 | n.d. | n.d. |
| 7/11 | 2 | 90 | No | – | – | – | – | – | – | – |
The pH of the reaction mixture was monitored and it was found that when the mixture containing palatinose (1), phosphoric acid, sodium tungstate and hydrogen peroxide was placed at 90°C, the pH, initially placed at 4, decreased rapidly during the course of the reaction. Thus, over the first 5 h, pH needed to be adjusted frequently (every 15 min) by addition of small portions of a molar solution of sodium hydroxide. After 5 h the pH levelled off and the reaction was continued for the next 20 h. An analysis of the main side products observed after acetylation showed that a α/β mixture of peracetylated glucose (4) was found when the pH was not carefully controlled (Scheme 2). When the reaction was indeed performed at pH 2, up to 8% yield of peracetylated glucose could be isolated and the CMGL (3) yield decreased to 12% showing poor stability of the glycosidic linkage at pH lower than pH 4 (Table 1, entry 2).
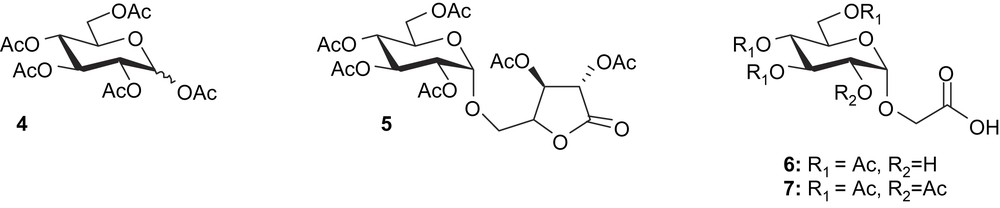
Control of the temperature proved to be a very important factor for improving the yield of CMGL, since when the reaction was conducted at 80 °C (Table 1, entries 2 and 3) another side product (5) was isolated and characterised. This compound is the result of an incomplete oxidation of the furanose ring of palatinose, which could give compound 5 after acetylation (Scheme 2). Interestingly, only trace amounts of this compound could be identified by TLC when the reaction was performed at 90 °C (Table 1, entry 1), showing that temperature is critical for obtaining a more complete oxidation of the fructose moiety in those conditions.
The large excess of hydrogen peroxide needed to be quenched prior to the acetylating step using either manganese dioxide or sodium sulfite. The use of catalytic amounts of MnO2 allowed the dismutation of H2O2, reducing the quantity of salts obtained at the end of the process. Thus, it should be considered as a good method for obtaining the CMG (2). However, it turned out to give a glassy residue after evaporation, which was found to be a problem for the following acetylation step, since it was very difficult to dissolve in a pyridine/acetic anhydride mixture. Despite the large quantities of salt obtained after evaporation, the use of sodium sulfite proved to be a more convenient way for the synthesis of CMGL (3).
According to the TLC analysis, the oxidative step giving access to CMG (2) was very reproducible, but the yields obtained of CMGL (3) were more sensitive to other parameters; hence we focused on the acetylation conditions and the stability of 3, which could explain the observed variations in yields. After oxidation, quenching of H2O2 and neutralisation, the initial workup involved a dilution with water and the diluted solution was freeze-dried to give a white powder which was subjected to acetylation [8]. In order to develop a robust and straightforward access to CMGL, we developed a direct protocol which did not involve the use of a freeze-drying step. Instead, after oxidation the obtained solution was directly evaporated and the residue was subjected to two different acetylating systems: acetic anhydride/pyridine or acetic anhydride/DMF/Et3N (Table 1, entries 1 and 5). Both conditions gave similar results in terms of yields and in both cases side products arising from the competitive opening of the lactone by traces of water were observed and identified as compounds 6 and 7. Hydrolysis of lactone 3 to give compound 6 was followed by NMR in a THF-d8/D2O mixture and was found to be complete after 4 weeks (Fig. 1). NMR experiments did not show any additional peaks to those expected for compound 6.
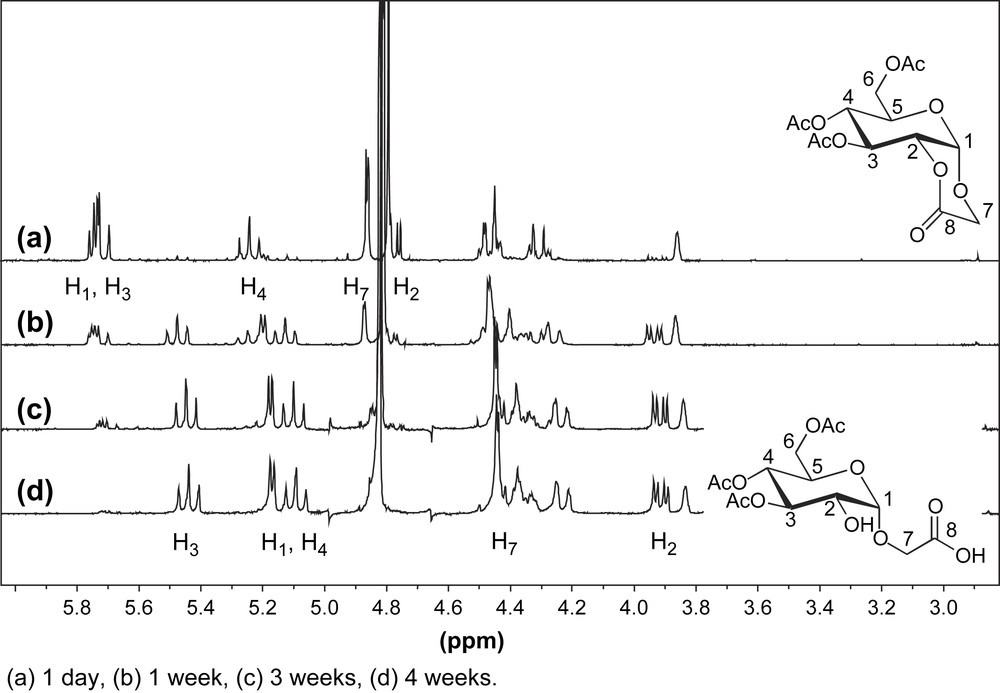
1H NMR spectra of the hydrolysis of lactone 3 in THF-d8/D2O. (a) 1 day, (b) 1 week, (c) 3 weeks, (d) 4 weeks.
Strict anhydrous conditions are therefore required in order to minimize the amount of side products formed during the acetylation step. Thus, the residue was placed under reduced pressure in a desiccator over P2O5 for 15 h to give access to a white solid on which the acetylation was then achieved.
Another side product, compound 7, was observed and its formation is explained by the acetylation of position 2 that has made impossible the lactone ring formation. It was shown that when compound 6 was indeed placed in an acetic anhydride/pyridine, lactone 3 was formed in quantitative yield, whereas when a small quantity of water was added (pyridine/Ac2O/H2O 50/10/1), CMGL (3) and compound 7 were obtained with, respectively, 58% and 23% yields.
Hydrolysis of lactone 3 to give 6 was also observed during the purification on silica gel column chromatography and was even faster when a small quantity of Et3N was added to the eluent mixture. Thus, after acetylation, the residue was washed with an ammonium chloride solution and carefully dried over sodium sulfate before being subjected to silica gel column chromatography. The obtained yellow oil was then precipitated to give 3 as a white solid which could be stored at 4 °C for several months.
For stability reasons and for widening the chemistry potentialities of α-d-glucopyranoside 2-O-lactone, similar structures bearing other protecting groups were synthesised by treatment of compound 2 with chloroacetyl chloride, pivaloyl anhydride or benzoyl chloride to give access to, respectively, 8 [2], 9 and 10 (Scheme 3).
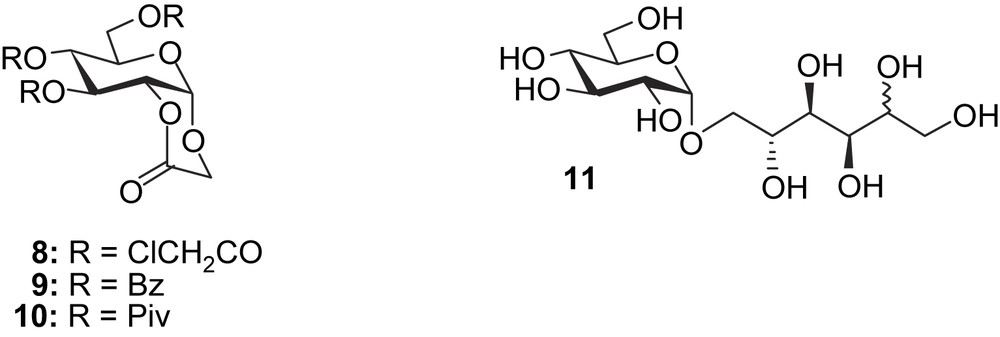
We also considered the use of isomalt® (11) as starting material (Scheme 3). Unlike what was observed for isomaltulose [1], no CMG (2) was detected by TLC when the reaction was carried out in the absence of sodium tungstate (Table 1, entry 7). Placed in the same condition used in the case of isomaltulose (pH 4, 90 °C), the reaction turned out to be very slow since it required 100 h and addition of 24 more equivalents of H2O2 to observe the complete disappearance of the starting material (Table 1, entry 5). Reaction was thus achieved at pH 2, giving access after 20 h at 90 °C to CMGL (3) with 16% yield (Table 1, entry 6).
In conclusion, this detailed study on the oxidative degradation of two different disaccharides followed by a direct acetylation has highlighted some key points of the sequence leading to carboxymethyl glucoside lactones. Even though the yields remain modest, these synthons are obtained through a straightforward and reliable process, which is competitive compared to multistep processes involving Koenigs–Knorr-type reactions.
3 Experimental part
All chemicals were purchased from Aldrich. Organic solutions were dried over anhydrous sodium sulfate. The reactions were monitored by thin-layer chromatography on silica gel 60 F254 (Merck); detection was carried out by charring with a 5% H2SO4 solution in ethanol. Silica gel (Kieselgel 60, 70–230 mesh ASTM, Merck) was used for flash chromatography. NMR spectra were recorded with a Bruker ALS300, DRX300 or DRX500 spectrometer in CDCl3. The signal of the residual protonated solvent was taken as reference. Chemical shift (δ) and coupling constants (J) are reported in ppm and Hz, respectively. Optical rotations were measured with a PerkinElmer 241 polarimeter for solutions in CH2Cl2 at room temperature. Elemental analyses were performed by ‘Service central de microanalyses du CNRS’, 69360 Solaize (France).
3.1 Synthesis of compounds 3, 5 and 6
3.1.1 Typical procedure for the synthesis of compound 3 (see Table 1 for variations in the conditions)
In a 500-mL three-necked flask equipped with a magnetic stirring bar, a reflux condenser and a pH meter probe, was placed a mixture of isomaltulose (1, 10 g, 29.2 mmol) in a (30% w/w) aqueous hydrogen peroxide solution (60 mL, 572 mmol). Sodium tungstate (2.32 g, 7.1 mmol) and 85% phosphoric acid (0.24 mL, 3.5 mmol) were added and the solution was adjusted to pH 4 using sodium hydroxide (1 M) before being placed at 90 °C. The pH was carefully adjusted every 15 min for the first 5 h and the mixture was stirred at 90 °C for 20 h. The solution was cooled to room temperature and potassium iodide (20 mg) was added to follow the quenching of the hydrogen peroxide excess. The medium was adjusted and maintained to pH 8 with sodium hydroxide (1 M), while an aqueous solution of sodium sulfite (10% w/v) was added until complete disappearance of the brown color. Solvents were evaporated under reduced pressure and the residue was placed in a desiccator over P2O5 for 15 h. The obtained yellow solid was then placed at r.t. in anhydrous pyridine (100 mL), acetic anhydride (75 mL) was added and the mixture was stirred for 24 h. Solvents were then removed under reduced pressure and the oily residue was taken up in CH2Cl2 (200 mL) and washed using an aqueous NH4Cl solution (10% w/v, 100 mL). The aqueous phase was extracted with CH2Cl2, the organic layers were combined, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography using a pentane/EtOAc (1/1) and the obtained yellow syrup was dissolved in CH2Cl2 (3 mL) and Et2O (60 mL), under stirring; pentane (150 mL) was slowly added and, after filtration, 3 was obtained as a white solid (2.33 g, 6.7 mmol, 23%).
3.1.2 Compound 5
Compound 5, a white solid, was isolated by column chromatography during the purification of compound 3. 1H NMR (500 MHz, CDCl3): δ 1.97 (s, 3H), 2.01 (s, 3H), 2.03 (s, 3H), 2.06 (s, 3H), 2.12 (s, 3H), 2.19 (s, 3H), 3.76 (dd, 1H, J = 11.5, J = 2.7 Hz), 4.02 (dd, 1H, J = 11.5, J = 3.9 Hz), 4.05–4.14 (m, 2H), 4.25 (dd, 1H, J = 12.3, J = 4.2 Hz), 4.49 (ddd, 1H, J = 6.3, J = 3.9, J = 2.7 Hz), 4.87 (dd, 1H, J = 10, J = 3.8 Hz), 5.05 (t, 1H, J = 9.8 Hz), 5.10 (d, 1H, J = 3.8 Hz), 5.42 (t, 1H, J = 10 Hz), 5.46 (t, 1H, J = 6.3 Hz), 5.65 (d, 1H, J = 6.3 Hz); 13C NMR (125 MHz, CDCl3): 20.4, 20.6, 20.7, 20.8, 20.8, 20.8, 61.7, 67.0, 67.7, 68.4, 70.1, 70.5, 72.0, 73.2, 79.0, 96.5, 168.6, 169.7, 169.8, 170.1, 170.2, 170.5, 170.8; [α]d20 +94 (c 1.0, CH2Cl2); HRMS (LSIMS) calcd for [M + H]+ 563.1612, found 563.1614; Anal. calcd for C14H18O10: C, 49.11; H, 5.38. Found: C, 49.06; H, 5.51.
3.1.3 Compound 6
Compound 6 was identified when lactone 3 was treated with a THF-d8/D2O mixture as shown in Fig. 1. 1H NMR (500 MHz, THF-d8/D2O): δ 2.06 (s, 3H), 2.09 (s, 6H), 3.80 (dd, 1H), 4.10 (m, 1H), 4.21 (m, 1H), 4.27–4.35 (m, 3H), 4.96 (t, 1H, J = 9.8 Hz), 5.04 (d, 1H, J = 3.0 Hz), 5.30 (t, 1H, J = 9.6 Hz); 13C NMR (125 MHz, THF-d8/D2O): 20.5, 21.5, 21.7, 63.5, 66.9, 69.2, 70.1, 70.9, 74.6, 100.5, 173.4, 174.3, 174.3, 174.4, HRMS (LSIMS) calcd for [M + Na]+ 387.0903, found 387.0905.
3.2 Synthesis of compounds 9 and 10
3.2.1 Compound 9
A solution of triethylammonium salt of CMG (2, 352 mg, 1 mmol) in anhydrous pyridine (20 mL) was placed at 0 °C. Benzoyl chloride (2.6 mL, 22.4 mmol) was added and the mixture was stirred for 3 days at room temperature. Solvents were evaporated under reduced pressure and coevaporated with toluene. The obtained residue was dissolved in CH2Cl2 and washed with a diluted solution of sodium hydrogenocarbonate. The organic phase was washed with brine and dried over sodium sulfate. The solvent was evaporated under reduced pressure and the obtained residue was chromatographed on silica gel using an hexane/ethyl acetate mixture (1:1) to give compound 9 as a white solid (160 mg, 30%). 1H NMR (300 MHz, CDCl3): δ 4.45 (dd, 1H, J = 12.9, J = 6.1 Hz), 4.55–4.70 (m, 4H), 4.77 (d, 1H, J = 18 Hz), 5.45 (d, 1H, J = 2.9 Hz), 5.62 (t, 1H, J = 9.6 Hz), 6.06 (t, 1H, J = 9.5 Hz), 7.3–7.6 (m, 9H), 7.8–8.1 (m, 6H); 13C NMR (125 MHz, CDCl3): 62.1, 64.4, 67.7, 70.5, 71.8, 76.2, 91.1, 163.4, 165.0, 165.6, 165.9; [α]D20 +87 (c 1.0, CH2Cl2); Anal. calcd for C14H18O10: C, 65.41; H, 4.54. Found: C, 65.24; H, 4.54.
3.2.2 Compound 10
A solution of triethylammonium salt of CMG (2, 1 g, 2.9 mmol) in anhydrous pyridine (30 mL) was placed at 0 °C. pivaloyl anhydride (60 mL) and DMAP (200 mg, 1.63 mmol) were added. The reaction mixture was stirred for 3 days at room temperature. Solvents were evaporated under reduced pressure and the obtained residue was chromatographed on silica gel using an CH2Cl2/acetone mixture (49:1) to give compound 10 as a white solid (873 mg, 63%). 1H NMR (300 MHz, CDCl3): δ 1.14 (s, 3H), 1.18 (s, 3H), 1.20 (s, 3H), 4.09–4.20 (m, 2H), 4.24–4.31 (m 1H), 4.40 (dd, 1H, J = 9.4, J = 3.0 Hz), 4.49 (d, 1H, J = 17.9 Hz), 4.68 (d, 1H, J = 17.9 Hz), 5.11 (t, 1H, J = 9.8 Hz), 5.32 (d, 1H, J = 3.0 Hz), 5.60 (t, 1H, J = 9.8 Hz); 13C NMR (75 MHz, CDCl3): 27.6, 27.7, 27.7, 39.4, 39.5, 39.5, 61.8, 65.0, 66.9, 71.3, 77.0, 91.7, 164.1, 177.1, 178.0, 178.3; [α]D20 +101 (c 1.0, CH2Cl2); Anal. calcd for C14H18O10: C, 58.5, H, 7.7. Found: C, 58.1; H, 7.8.
Acknowledgements
Financial support from CNRS, MENSER, and TEREOS is gratefully acknowledged. We thank Cargill SA for a gift of isomaltulose. ANRT, CNRS, and TEREOS are also acknowledged for grants to R.P., M.D. and S.T. The present address of Dr Trombotto is: Laboratoire d'ingénierie des matériaux polymères, CNRS UMR 5627, Université Claude-Bernard–Lyon-1, ISTIL, 43, bd du 11-Novembre-1918, 69622 Villeurbanne cedex, France.