

1 Introduction
Thioureas are molecules of intensive current interests with regard to their roles in hydrogen bonding-based receptors for anions [1] and organocatalysts in synthetic chemistry [2]. In order to improve the efficiency, their hydrogen bonding interactions with guest molecules are concerns. A straightforward method of enhancing hydrogen bonding for a given guest molecule is to increase the acidity of thioureido –NH protons. This can be done to a high extent until the occurrence of deprotonation of the acidic –NH protons in the presence of relatively basic guest. We have been exploring ways of enhancing the hydrogen bonding of thiourea-based receptors while the acidity of the thioureido –NH protons remains not that high [3]. We previously reported that with N-benzamido-N′-arylthioureas (1, Fig. 1) the anion binding constants in acetonitrile were much higher than those of the classic N,N′-bisarylthioureas (2, Fig. 1) despite not higher acidity of the thioureido –NH protons in the former. In those reports we showed that the spectral signal reporter, i.e. benzamido moiety, was decoupled from the thiourea binding site by the twisted N–N single bond [4], suggesting that NHNH is an interesting spacer between the signal reporter and the binding site. We then extended our scope of research to N,N′-bis(benzamido)thioureas (3, Fig. 1) by examining them as anion receptors, hoping to expand the variety of thiourea-based receptors for anions and to provide possible structural hints for designing new thiourea-based organocatalysts.
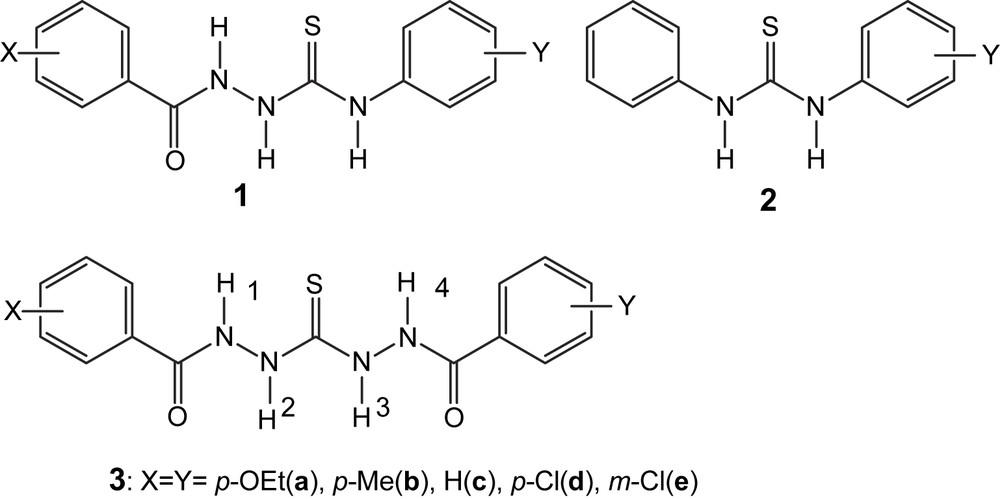
Chemical structure of thioureas and N-benzamidothioureas. Numbering of –NH protons was also given for 3.
2 Experimental
2.1 General procedures for preparation of N,N′-bis(benzamido)thioureas (3)
N,N′-Diaminothiourea (0.21 g, 2 mmol) was dissolved in aqueous NaOH solution (2 mL, 2 mol L−1), which was added dropwise into a solution of benzoyl chloride (0.56 g, 4 mmol) in tetrahydrofuran at 0–5 °C. The reaction mixture was then stirred at room temperature for 2 h to give a white precipitate. The as-obtained products were purified by repeated recrystallizations from ethyl acetate, 0.58 g, 72% for a; 0.51 g, 74% for b; 0.50 g, 80% for c; 0.54 g, 71% for d; and 0.59 g, 77% for e.
Compound 3a: mp 186–188 °C. 1H NMR (500 Hz, DMSO-d6) δ (ppm): 1.342 (t, J = 7 Hz, 6H), 4.095 (q, J = 7 Hz, 4H), 6.998 (d, J = 8.5 Hz, 4H), 7.894 (s, 4H), 9.665 (s, 1H), 10.092 (s, 1H), 10.262 (s, 1H), 10.381 (s, 1H). 13C NMR (125 MHz, DMSO-d6) δ (ppm): 14.537, 63.328, 113.894, 124.590, 125.071, 129.446, 129.921, 161.180, 164.896, 165.642, 183.795. HRMS: found m/z 403.1456; calcd for [C19H22N4O2S+] 403.1440. Compound 3b: mp 198–199 °C. 1H NMR (500 Hz, DMSO-d6) δ (ppm): 2.364 (s, 6H), 7.289 (d, J = 8 Hz, 4H), 7.827 (s, 4H), 9.704 (s, 1H), 10.124 (s, 1H), 10.339 (s, 1H), 10.459 (s, 1H). 13C NMR (125 MHz, DMSO-d6) δ (ppm): 21.028, 127.593, 128.023, 128.673, 128.821, 129.802, 130.267, 141.550, 141.719, 165.328, 166.034, 183.779. HRMS: found m/z 343.1237; calcd for [C17H18N4O2S+] 343.1229. Compound 3c: mp 192–193 °C. 1H NMR (500 Hz, DMSO-d6) δ (ppm): 7.491(t, J = 7 Hz, 4H), 7.570 (t, J = 7.5 Hz, 2H), 7.920 (s, 2H), 7.955 (s, 2H), 9.771 (s, 1H), 10.191 (s, 1H), 10.440 (s, 1H), 10.563 (s, 1H). 13C NMR (125 MHz, DMSO-d6) δ (ppm): 127.563, 127.981, 128.301, 131.735, 132.511, 132.929, 165.449, 166.051, 183.776. HRMS: found m/z 315.0908; calcd for [C15H14N4O2S+] 315.0916. Compound 3d: mp 268–269 °C. 1H NMR (500 Hz, DMSO-d6) δ (ppm): 7.580 (d, J = 8.5 Hz, 4H), 7.933 (s, 4H), 9.820 (s, 1H), 10.230 (s, 1H), 10.552 (s, 1H), 10.655 (s, 1H). 13C NMR (125 MHz, DMSO-d6) δ (ppm): 128.242, 128.453, 129.443, 129.880, 131.343, 131.687, 136.469, 136.578, 164.538, 165.133, 183.802. HRMS: found m/z 383.0132; calcd for [C15H12Cl2N4O2S+] 383.0136. Compound 3e: mp 217–218 °C. 1H NMR (500 Hz, DMSO-d6) δ (ppm): 7.542 (t, J = 8 Hz, 2H), 7.656 (d, J = 8 Hz, 2H), 7.870 (d, J = 5 Hz, 2H), 7.942 (s, 1H), 8.033 (s, 1H), 9.865 (s, 1H), 10.271 (s, 1H), 10.610 (s, 1H), 10.705 (s, 1H). 13C NMR (125 MHz, DMSO-d6) δ (ppm): 126.302, 127.372, 127.926, 130.225, 130.488, 131.566, 133.004, 133.201, 134.569, 134.981, 164.215, 164.865, 183.721. HRMS: found m/z 383.0120; calcd for [C15H12Cl2N4O2S+] 383.0136.
Absorption spectra were recorded on Varian Cary 300 absorption spectrophotometer. IR spectra were obtained with a Nicolet FT-IR 360 with a KBr pellet. NMR data were acquired on a Varian Unity+ 500 MHz or Bruker 400 MHz NMR spectrometer using TMS as an internal reference. HRMS were obtained by injection of a methanol solution of the sample on a Micromass-LCT high-resolution mass spectrometer. X-ray diffraction data of 3e were collected on a Bruker SMART APEX 2000 CCD diffractometer, working with a graphite-monochromated Mo Kα radiation (λ = 0.71073Å). The structure was solved by direct methods and refined on F2 by full-matrix least-square methods with the SHELX-97 package. All of the non-H atoms were refined anisotropically. The structural data were deposited in the Cambridge Crystallographic Data Centre under No. CCDC 630944. Solvents for spectral measurements were purified until no fluorescent impurity could be detected. Anions were made in their tetrabutylammonium salts from neutralization of the corresponding acids by tetrabutylammonium hydroxide.
3 Results and discussion
The NMR data, in particular those of the thioureido and amido –NH protons of 3a–e, were compared and correlated with the Hammett substituent constant (σx). It deserves pointing out that, despite the symmetric character of the molecules, the NMR signals in DMSO-d6 of the two thioureido and of the two amido –NH protons, respectively, were not the same, indicative of their different electronic environment (Fig. 2). Note that with N-benzamido-N′-phenylthioureas (1) under the same conditions, the two thioureido –NH protons had very close chemical shifts [3b]. Plotting –NH protons' chemical shifts of 3a–e against Hammett constant of the substituent X showed that linear correlations existed between them with varied slopes (Fig. 3). It was found that the linear slopes for two amido –NHs and two thioureido –NHs are, respectively, close, while the slopes for amido –NHs are much higher than those of thioureido –NHs. The latter were also much higher than those of N-benzamidothioureas (1) [3b]. This means that the electronic effect of substituent at benzamido phenyl ring in 3a–e could not be efficiently communicated to the thioureido –NH protons. This is in line with our previous conclusion made with N-amidothioureas (1) that the N–N single bond is twisted [3a,3b]. Crystal structure of 3e supported this assumption by indicating a clear twisting of the N–N single bond (Fig. 4). From these structural analyses, it appears that anion binding to the thiourea moiety in 3 would not disturb very much the electronic characters and consequently the absorption spectra of the benzamide chromophores in 3.
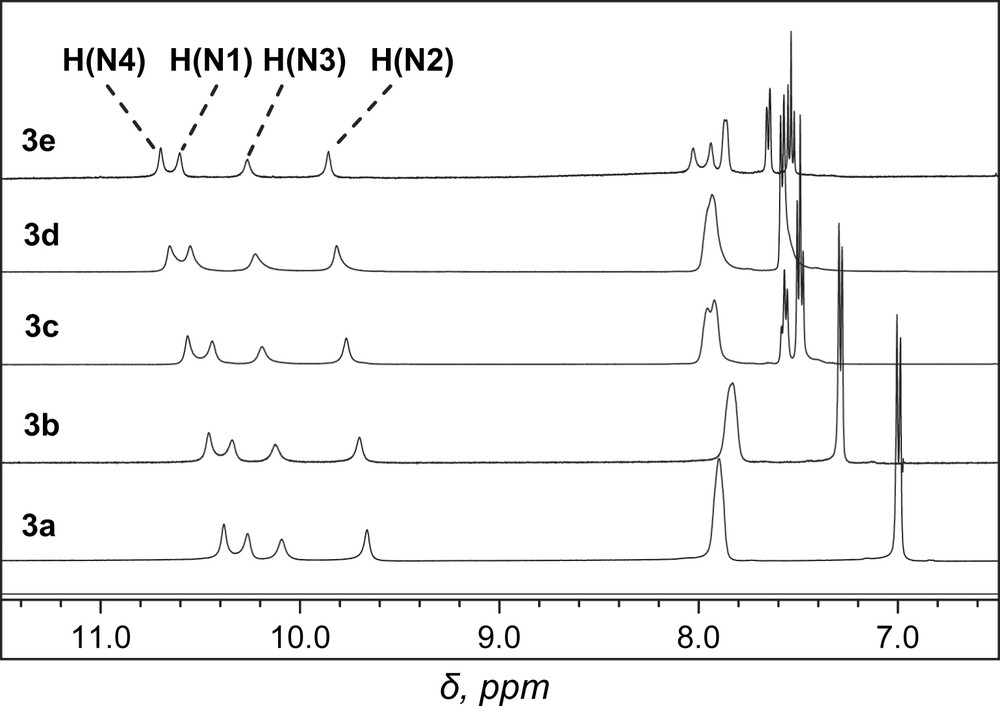
Portion of 1H NMR spectra of 3a–e in DMSO-d6. For numbering of –NH protons, see Fig. 1.
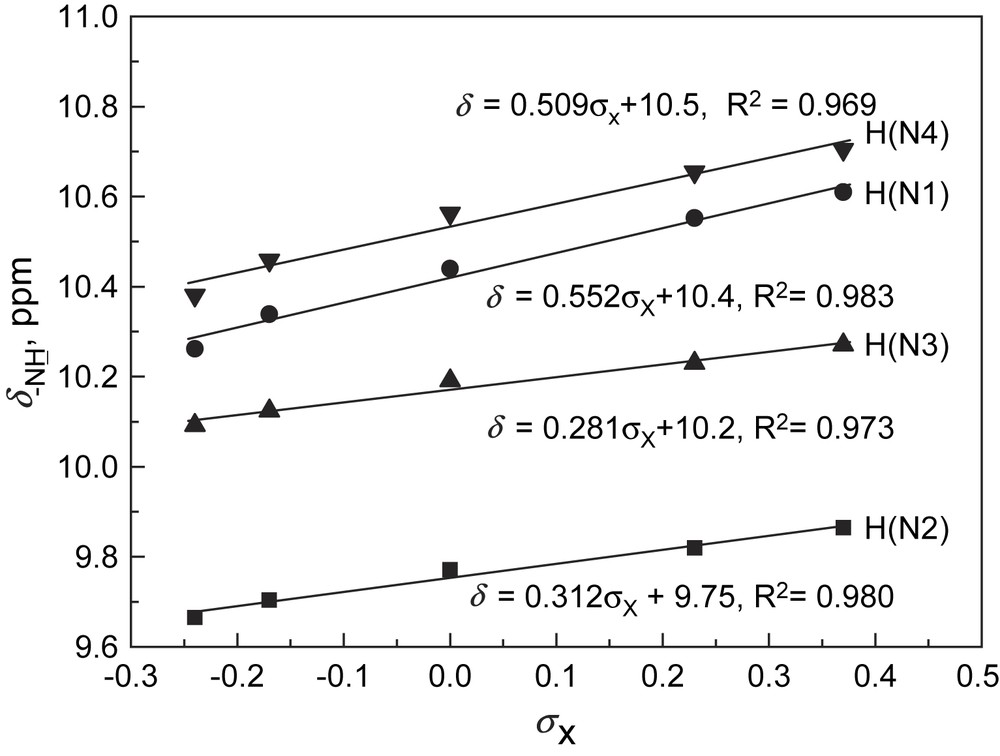
Chemical shifts of –NH protons in 3a–e in DMSO-d6 versus Hammett substituent constant. For numbering of –NH protons, see Fig. 1. The corresponding linear correlations for amido –NH and two thioureido –NHs in N-benzamidothioureas (1) are δ = 0.463σx + 10.5, δ = 0.0977σx + 9.80, δ = 0.128σx + 9.69, respectively [3b].

Crystal structure of 3e with DMSO solvent molecules. Symmetry codes: A, −x + 1, −y, −z + 1; crystal system and space group: triclinic, P¯1.
Absorption spectra of 3 monitored in acetonitrile (ACN) in the presence of anions such as F−, AcO−, and H2PO4−, however, indicated substantial changes. Fig. 5a shows the absorption titration traces of 3c by AcO− in ACN. Notably a new and lower energy absorption band was developed at the expense of the original absorption of the benzamide moiety, with two isosbestic points. Titration plots of the absorbance of the new band (Fig. 5b) point to a much higher sensitivity of 3c toward AcO−, F−, and H2PO4− compared to that of other anions tested. With other receptors, similar spectral variations and titration profiles were observed (Table 1). 1H NMR titrations confirmed the hydrogen-bonding interaction of receptors with anions. It was noted in Fig. 6 that the signals of thioureido –NHs of 3c were broadened and disappeared, whereas those of amido –NHs mixed and disappeared. Meanwhile the NMR signals of aromatic –CHs underwent upfield shifts. Job plot suggests a 1:1 stoichiometry in AcO− binding to 3c (Fig. 7). Non-linear fittings of the titration plots led to binding constants of anions to receptors (Table 1), which were found higher than those of N-benzamidothioureas (1) [3b]. In Table 1 it is also noted that anion binding induced spectral red shifts are more substantial than those observed with 1 [3b]. Plot of the absorption energy of the new band of receptors 3a–e in the presence of AcO− against Hammett constant of the substituent X was found linear, with a slope of −0.361 eV (Fig. 8). This means that the new absorption band is of charge-transfer (CT) nature [5], with benzamide chromophore being the electron acceptor (A), while the anion-thiourea-binding unit the donor (D). The anion-binding complexes of 3a–e would then be in an “A–D–A” structural framework [6]. Compared to the corresponding correlation of 1 (hν = 3.67–0.337σx) [3b], it was found that the CT absorption of 3–AcO− complexes appeared at higher energy than that of 1–AcO− complexes and showed stronger substituent dependence. The latter is also indicative of a more substantial charge transfer in the anion-binding complexes of 3 [5]. The fact that the absorption spectra of 3a–e underwent change upon anion binding and the occurrence of charge transfer in the binding complexes suggest a conformation change in the N–N single bond upon anion binding that allows an efficient electronic communication between benzamide and thiourea moieties in the binding complexes and a substantial increase in the electron donating ability of the anion-thiourea binding unit. It is of interest to indicate that upon charge transfer in the binding complexes a positive charge will be developed in the thiourea moiety that is in turn good for anion binding, which means a positive feedback to the binding of anion. This was supported by the fact that the anion-binding constants of 3 were higher than those of 1. The binding constants of 3a–e for F− and AcO− in ACN are too high to allow a credible correlation with the Hammett constant of substituent Y. Those of H2PO4−, however, was found to show a stronger dependence (ln K = 10.85 + 3.27σx, γ = 0.923) than those of 2 and in particular of 1 (ln K = 10.21 + 2.07σx, γ = 0.756) [3a]. This observation serves as another support for the assumed positive feedback to anion binding of the anion-binding-induced conformation change in N–N bond and charge transfer, defining 3 hence as allosteric receptors for anions [7].
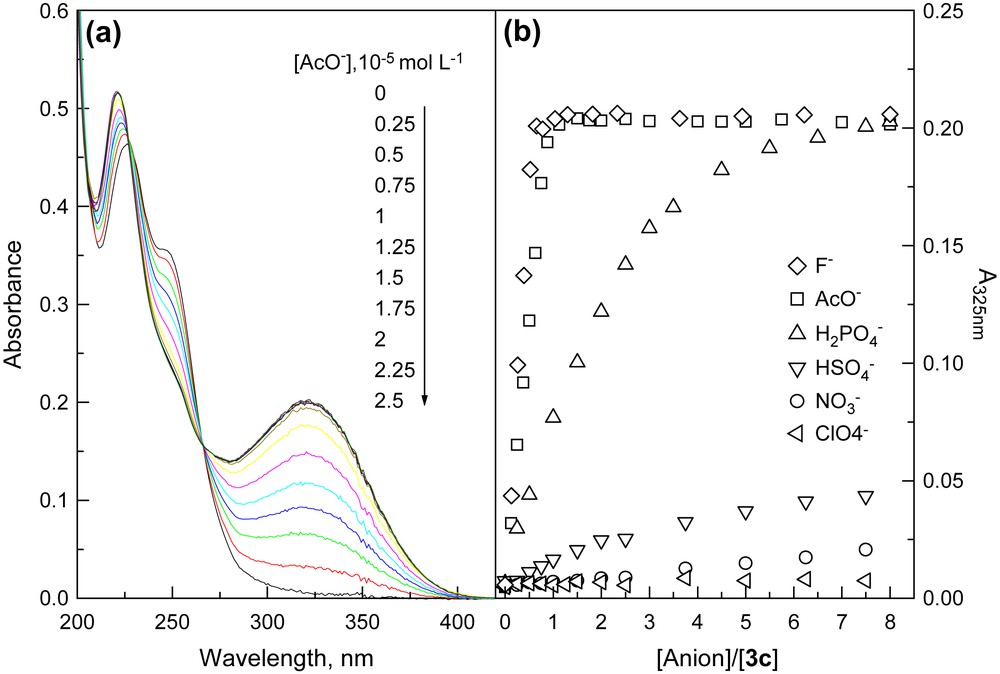
(a) Absorption spectra of 3c in acetonitrile in the presence of acetate anion and (b) plots of absorbance of the new band at 325 nm against anion concentration.
Absorption and anion binding parameters of receptors 3a–e in acetonitrile
| λmax (R)a, nm | ɛb, 104 L mol−1 cm−1 | λmax (R-AcO−)c, nm | Δhνd, cm−1 | Kae, 106 mol−1 L | |||
| AcO− | F− | H2PO4− | |||||
| 3a | 256 | 5.02 | 320 | 7013 | 9.94 | >10 | 0.0152 |
| 3b | 242 | 2.95 | 321 | 10,170 | >10 | >10 | 0.0382 |
| 3c | 227 | 2.23 | 325 | 9391 | >10 | >10 | 0.0803 |
| 3d | 240 | 2.94 | 335 | 11,716 | >10 | >10 | 0.1213 |
| 3e | 249 | 1.61 | 338 | 10,575 | >10 | >10 | 0.132 |
a Absorption maximum of receptor.
b Molar absorption coefficient.
c Absorption maximum of receptor–AcO− complex.
d Absorption shift induced by AcO− binding.
e Anion binding constant.
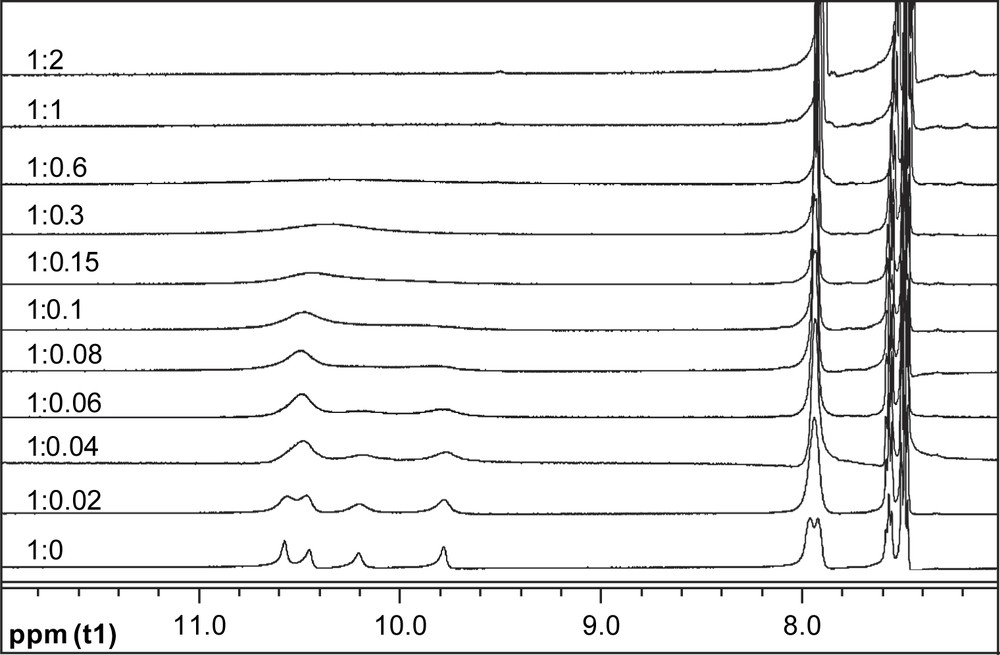
Portion of 1H NMR spectra of 3c in DMSO-d6 in the presence of AcO− of increasing equivalent.
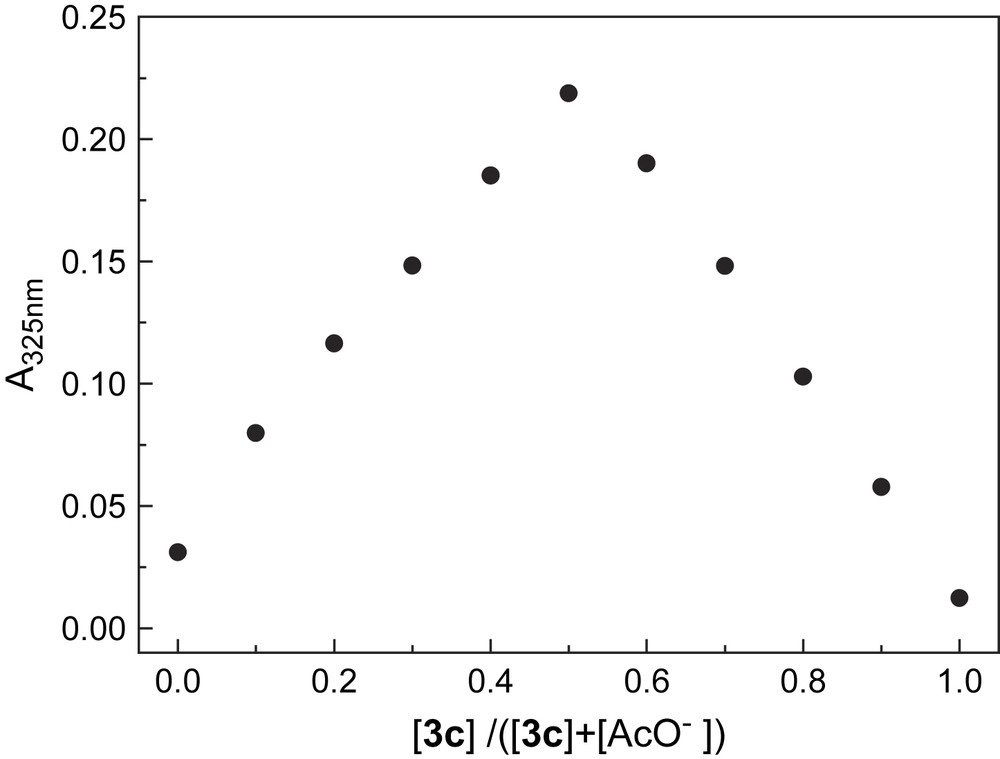
Job plot for binding of AcO− to 3c in acetonitrile by monitoring absorbance at 325 nm. Total concentration of AcO− and 3c is 4.0 × 10−5 mol L−1.
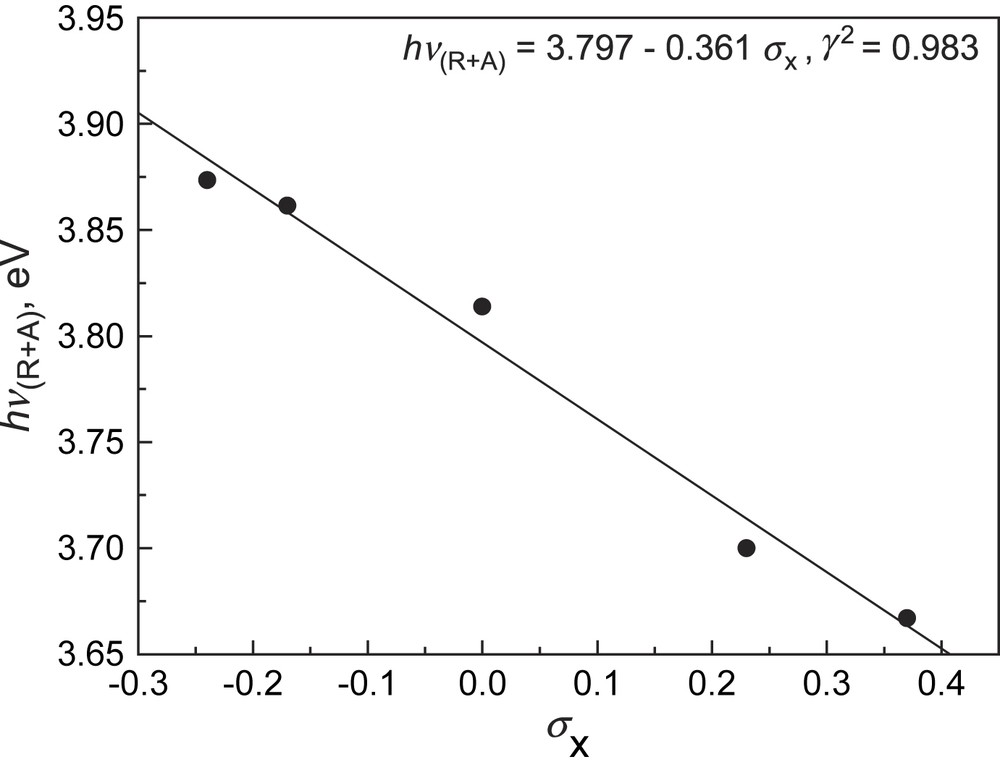
Linear correlation of absorption energy of receptor–AcO− binding complex in acetonitrile with Hammett substituent constant. The corresponding linear correlation with 1 was hν = 3.67–0.337σx [3b].
4 Conclusions
The symmetric N,N′-bis(benzamido)thioureas (3) were found much better in binding anions in acetonitrile with binding constants for F− and AcO− at 107 M−1 or higher orders of magnitude and to show more substantial absorption spectral red-shifts, compared to N-benzamidothioureas (1). The N–N single bond in 3 was found twisted that actually stopped the electronic communication of benzamide chromophore and thiourea binding site, which means that no absorption spectral change could be expected. The acidity of the thioureido –NH protons was shown to be lower than that in 1 and 2. The substantial response to anions of absorption spectra of 3 therefore indicated a conformation change in the N–N bond that eventually switches on intramolecular charge transfer in the anion-binding complex. The charge-transfer absorption of the anion binding complexes of 3a–e showed a stronger dependence on the substituent constant, pointing to a higher degree of charge transfer in the complexes, which explains the observed stronger substituent dependence of the anion binding constant. The reported thiourea-based receptors are hence defined as allosteric receptors with higher anion binding constants, while the acidity of thioureido –NH protons is lower. As in the anion binding complexes of 3a–e, an “A–D–A” structural framework is generated, an extension of generating “D–A–D”-type symmetric derivatives is underway.
Acknowledgments
This project has been supported by the National Natural Science Foundation of China through Grant Nos. 20425518 and 20675069, the Ministry of Education of (MOE) China, and the VolkswagenStiftung through Grant No. I/77 072.