

1 Introduction
Among the basic principles of Green Chemistry, one is to develop the use of renewable starting materials as an alternative to fossil resources. Carbohydrates are, in this respect, a considerable reserve of carbon-containing molecules, and this is why they have attracted continual interest in the past decades for the synthesis of a variety of compounds, such as surfactants or polymerizable derivatives. This is true for polysaccharides, such as starch, cellulose, chitin, inulin, and also for smaller carbohydrates, such as glucose and sucrose [1–4]. However, carbohydrates are complex molecules with a complex chemistry, because of their moderate stability to heat or to acidic or basic conditions, and because of their polyfunctionality. The recent progresses in the chemistry of carbohydrates are essentially connected with complex targets of high added value or of high biological interest, and are based on subtle protection–deprotection strategies and the use of fancy catalysts and reagents. The impressive level of efficiency reached in this chemistry has provided fundamental clues towards the understanding of many biological processes in which carbohydrates are involved and therefore opened new chapters of biochemistry and therapeutics based on glycoconjugates. Parallel to this, some basic research is still necessary, notably aiming at simplifying the multi-step routes, decreasing the use of protective groups, and finding new direct and simple processes for the chemical transformation of simple carbohydrates to low-added value derivatives. Sucrose, the most abundant pure organic molecule produced at the industrial scale (>125 Mt/yr) from renewable resources (sugar cane and sugar beet), is, in this respect, a fascinating example. Its chemistry is typical of the difficulty to derivatise simple carbohydrates [5–9]. Sucrose (1) is a disaccharide constructed with a glucose moiety and a fructose one connected at their anomeric carbon atoms (Scheme 1). It therefore does not contain a hemiacetalic linkage and is, hence, a ‘non-reducing’ sugar. The absence of masked carbonyl function essentially limits its chemistry to that of the numerous (three primary and five secondary) hydroxyl groups. Many products can arise from a simple reaction, resulting from different degrees of substitution (mono-, di-, etc.) and from different positions of the substituents on the disaccharidic skeleton. It is obvious that the control of the selectivity of the reactions is the central issue if direct reaction of sucrose is desired. As a simple example, the physicochemical properties of a monosubstituted sucrose fatty acid ester and of the corresponding disubstituted one are reverse, the first being an oil-in-water emulsifier and the second a water-in-oil one: two different products, two different physicochemical properties, two different industrial applications [10–13]. The degree of substitution is therefore one key parameter. It is more difficult to establish such an importance for the structural position at one or the other of the hydroxyl groups of sucrose, and it was only recently demonstrated that indeed, this could also be a critical parameter in some cases [14]. Whatever functional consequences it has on the product properties, the regioselectivity can be seen as the best source of control of degree of substitution. Indeed, when one of the eight hydroxyl groups exhibits a preeminent reactivity towards an electrophilic species, the second substitution occurs in a slower manner, allowing us to trap some low substituted derivatives. The regioselectivity, in the case of reactions under kinetic control, is the result of a chemoselectivity of one alcohol function among the others, and can possibly be controlled by structural and electronic factors.
The purpose of this review, by combining recent work of our group and literature data, is to show the connections between the structure of sucrose and its reactivity. New direct routes and new reaction conditions for the preparation of functional derivatives of sucrose are described. Notably, it is demonstrated that the classically accepted view of sucrose nucleophilic reactivity describing the primary OH-6 and 6' as more reactive positions is not valid anymore, since depending on the nature of the electrophilic partner and on the reaction conditions and catalysts, the outcome of the transformation could be oriented towards many different positions, often with reasonable control of selectivity.
The issue of the solvent, also a key element with respect to green chemistry, has to be addressed in the special case of the chemistry of sucrose, which is soluble to a reasonable extent only in water and polar aprotic solvents such as DMF and DMSO. Interestingly, the very same causes that give OH-2 a special acidity and reactivity also makes possible the use of aqueous or mixed aqueous–organic media, even in the presence of water-sensitive electrophilic species.
2 Etherification of sucrose
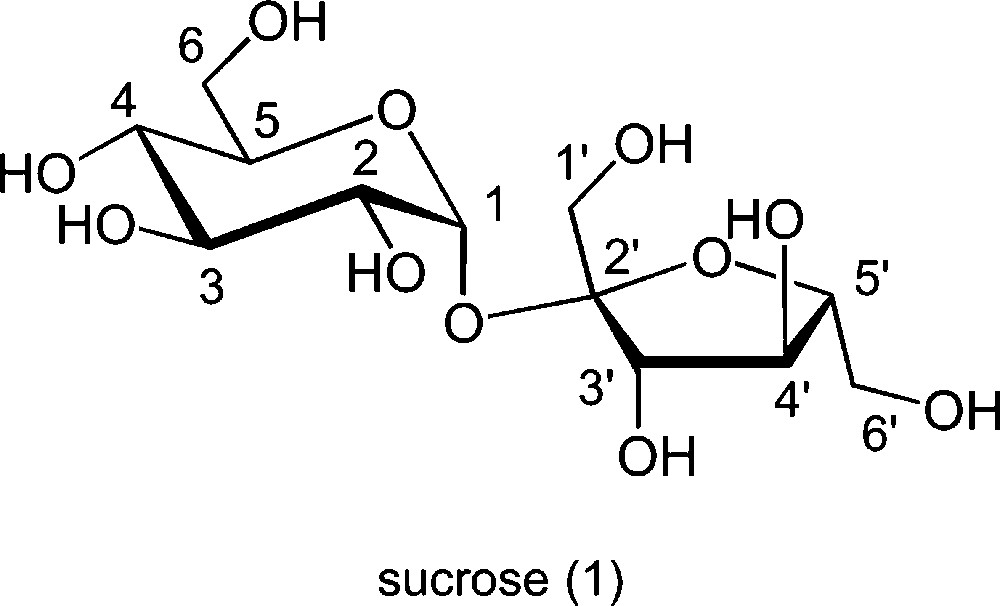
The chemical stability of the ether linkage, preventing reversibility, allows a direct connection between the distribution of the products and the relative kinetic reactivity of the competitive nucleophilic centres. Thus, etherification is the ideal reaction for studying the relative reactivity of the eight hydroxyl groups of sucrose, with the goal of associating intrinsic conformational and electronic properties with possible observed selectivities. The conformation of sucrose is mostly due to intramolecular hydrogen bonds present in the solid state (between OH-6′ and the pyranosidic oxygen atom of the glucose moiety, and between OH-1′ and O-2). In solution, only one of these bonds remains, involving O-2 and either OH-1′ or OH-3′ (Scheme 2), leading to an conformational equilibrium (A and B) that has been rationalised by spectroscopic and theoretical studies [15–19]. These three positions are those that are also close to the anomeric centres. Direct consequences on the nucleophilic behaviour of sucrose have been observed at these positions, and connected with the relative acidity of the OH groups [20]. For example, 2-O-benzyl sucrose (2) is obtained in 80% ratio among the monosubstituted esters besides 1′ and 3′ esters, by reaction in DMF with benzyl bromide in the presence of sodium hydride [21]. Also, electrochemical etherification of sucrose led to methyl ethers essentially at 2′ (major) and 1′ and 3′ positions [22]. On the other hand, when large substituents are involved, the reaction takes place at the primary OH-6 and OH-6′, and possibly at the sterically more hindered OH-1′, as in the case of trityl (3) or bulky silyl ethers [23–26]. The orientation of the reaction towards derivatives substituted at OH-2 and OH-1′ could be also observed in aqueous medium.
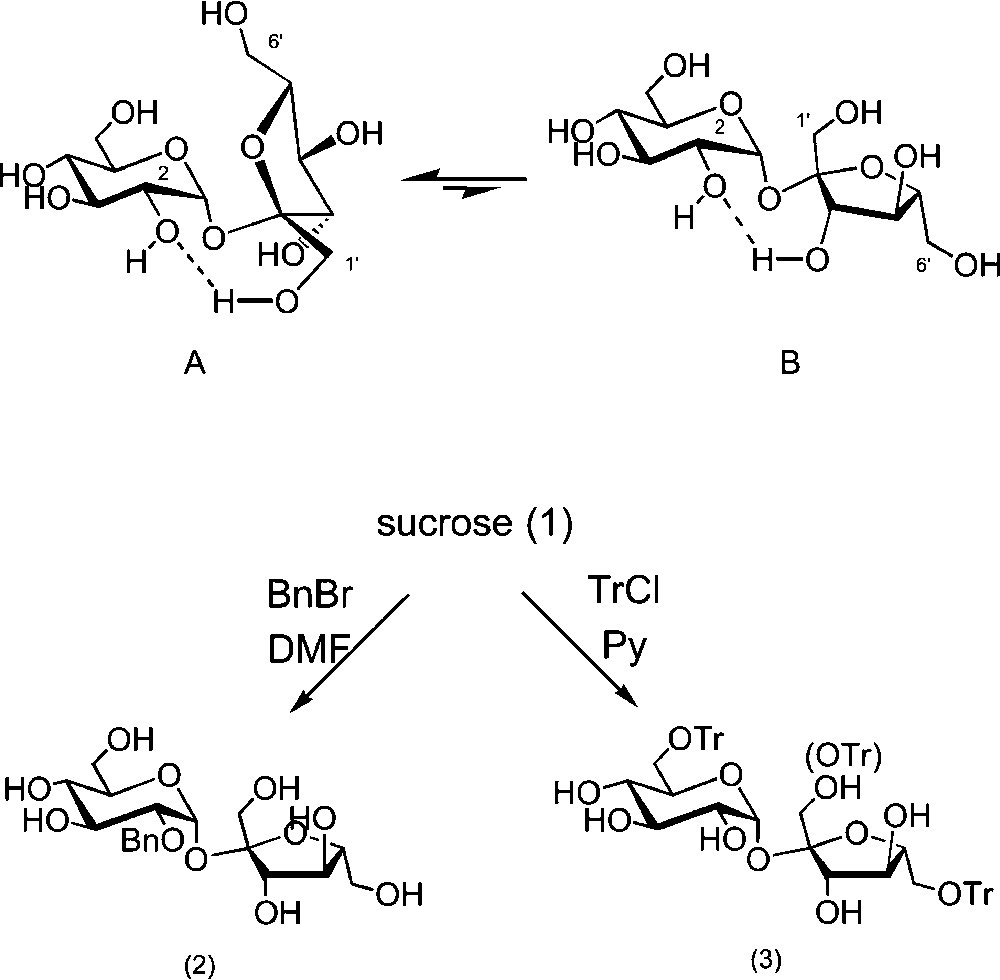
Amphiphilic sucrose hydroxyalkyl ethers are formed by reaction with fatty epoxides in a slightly less regioselective manner compared to DMSO, but the same major isomers are formed [27]. This shows that the solvation of sucrose has only a little influence on the relative reactivity of the hydroxyl groups, consistent with the spectroscopic and theoretical studies that have demonstrated the presence of a remaining hydrogen bond involving OH-2 and OH-1′ through a bridging water molecule [28,29]. In this addition of sucrose on fatty epoxides, the main obstacle was the heterogeneity of the medium, imposing to adapt the nature of the basic catalyst (notably tertiary amines) and possibly the presence of surfactant additive [30]. The desired monosubstituted hydroxyalkyl ethers 4a–h were obtained and the regioisomers could be separated, allowing a detailed study of the influence of the position of the substitution on their liquid-crystal properties (Scheme 3). Thereby, the global shape of the hydrophilic moiety of the molecule is essential in the thermotropic packing of these materials, and, notably, the substitution at OH-2 or OH-3′, which prevents the usual intramolecular hydrogen bond network from being established, exhibits a different thermotropic behaviour (colomnar) as compared to other isomers (smectic A) [14]. These results are consistent with the conformational study of 2-O-lauroyl sucrose, for which the fatty chain adopts an arrangement perpendicular to disaccharidic backbone [31].
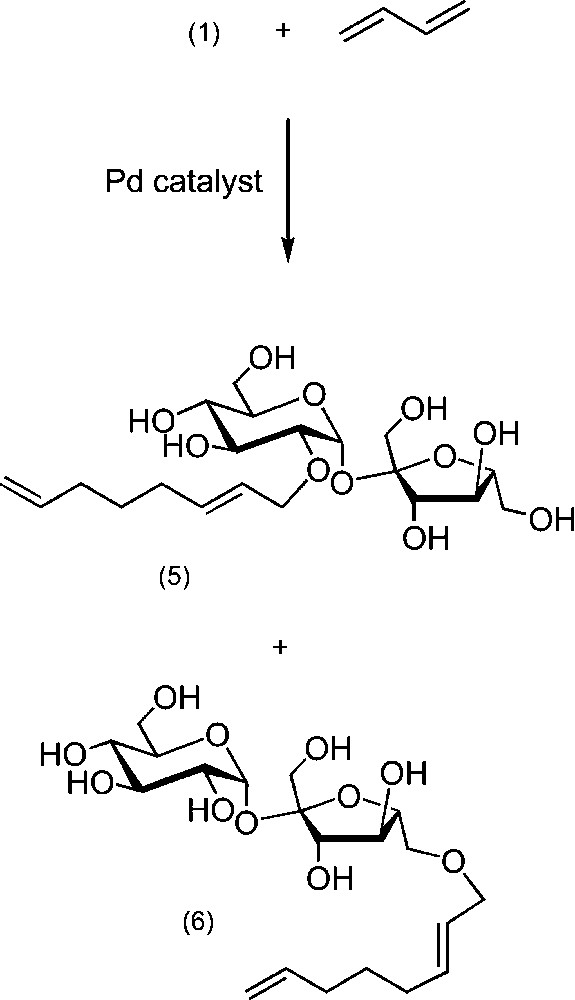
Carbohydrate ethers can also be obtained by palladium-catalysed telomerisation of butadiene [32–37]. With respect to the green context, this a very interesting alternative, though less selective, for the preparation of sucrose octadienyl ethers (isomers 5 and 6 as major products), which can be achieved in aqueous medium by using sulfonated phosphines as water-soluble ligands (Scheme 4).
3 Esterification of sucrose
Sucrose esters have industrial interest in the domain of surfactants, bleaching boosters and fat substitutes. Polysubstituted fatty acid esters have the similar properties as those of triglycerides, but are not substrates for digesting lipases [38–39]. They can therefore be used as non-caloric fats and are actually produced for this application in Northern America (Olestra®). Some polysubstituted sucrose isovalerates were also found to be interesting candidates as scaffold for multidrug resistance modulators [40]. On the other hand, low substituted fatty acid esters are food and cosmetic emulsifiers that are produced on an industrial scale [10–13,41–44]. They are used as mixtures of regio-isomers as well as mono-, di- and triesters, with physicochemical properties depending on the average degree of substitution and on the length of the fatty chains. However, the precise behaviour was shown to depend also on other factors, such as the presence of small amounts of remaining fatty acid or soap. This prevents to use the too simple commercial HLB value to really foresee their emulsifying properties. The study of Winsor systems was shown to be an interesting alternative in this respect [45]. In order to get some precise structure–activity relationships in this series of compounds, it is necessary to prepare pure sucrose fatty esters of defined structure. Consequently, many studies aiming at more direct or selective syntheses have been reported. But unlike ethers, esters are unstable, stepwise acyl group migrations from one position to another being common. Esterifications and transesterifications are usually conducted under basic catalysis, and lead essentially to derivatives substituted at the primary positions. As mentioned above, it does not necessarily translate the relative kinetic reactivity of the hydroxyl groups, since migrations are also catalysed by bases. However, selective acylation at OH-2 has been shown to produce ester 7 using N-acylthiazolidine thiones as activated acylating agent (Scheme 5) [46]. The selective preparation of 2-O-tosyl sucrose was also recently described [47]. In precise basic conditions, the 2-O-ester can be further transformed by controlled migration. This provides a method for the preparation of 6-O-acyl sucrose derivatives [48,49]. Esters at OH-6′ can also be obtained via a chemoenzymatic sequence [50]. Other methods involving metal complexes led to derivatives substituted at secondary positions 3 or 3′ [51]. Substitution at primary positions can be rather selective, providing 6-O-esters (8), when using controlled amounts of acylating agents and low temperatures [52,53] of tin derivatives [54,55].
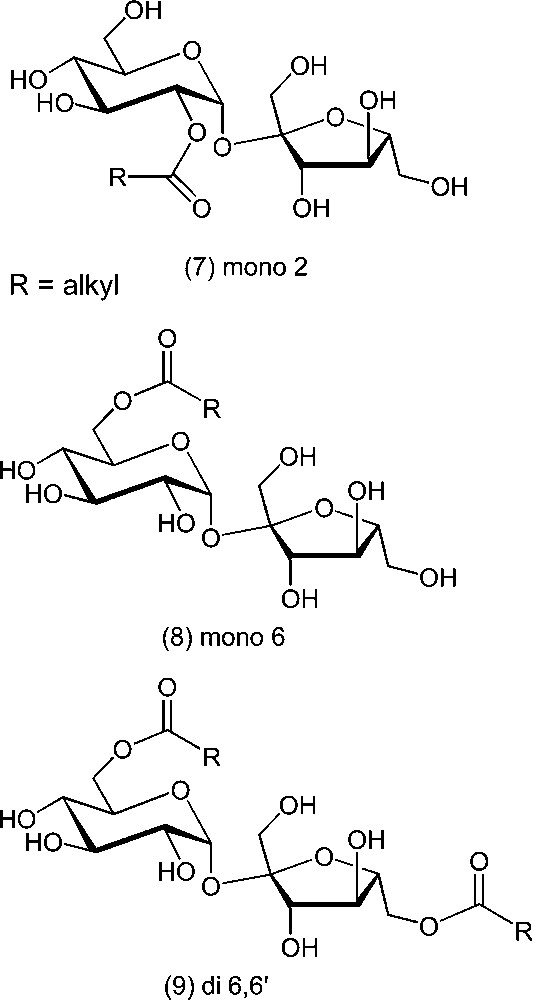
Under Mitsunobu conditions, the 6,6′-di-O-esters (9) are prepared, with possible trapping of the 6-O-ester 8, which appears to be formed faster [56,57]. Competitive formation of anhydro derivatives is observed in the case of less reactive carboxylic acids. Mixed disubstituted sucrose derivatives can be obtained from monofunctionalised substrates [58–60]. Phosphates or phosphonates esters were also prepared by the same route [61,62].
An interesting question is to know whether sucrose esters could be prepared in aqueous medium instead of organic solvent-based systems that might bring some additional costly purification steps. The study of the synthesis in water of fatty acid esters and carbonates by reaction with acid chlorides or chloroformates, respectively, revealed that, in the presence of base, good conversion of the acylating agents was obtained, due to a faster reaction of sucrose as compared to water [63,64]. However, in such conditions, hydrophobic-driven polysubstitution occurred predominantly, increasingly with longer fatty chains. Addition of DMAP, helping the fatty acid derivative to be less sensitive to the hydrophobic effects by the formation of an acyl ammonium intermediate salt, and use of water–isopropanol mixtures in order to decrease the strength of the water structure responsible for the hydrophobic effects, were shown to fight the polysubstitution tendency. Monosubstituted derivatives were thus obtained in good yield under such conditions. The regioselectivity of the reactions performed in aqueous media was also studied, showing the reaction to take place at some secondary positions, even though compounds are finally those having the primary positions acylated due to intramolecular migrations [65]. The formation of the ester at OH-2 was clearly shown to occur first, like in organic medium, despite the perturbation of the intramolecular hydrogen bond network due to solvation. These results are again consistent with the studies on the conformation of sucrose in water.
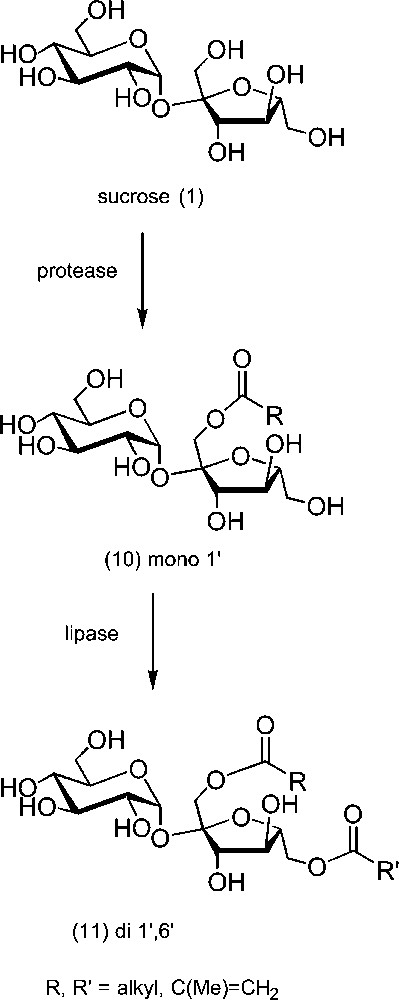
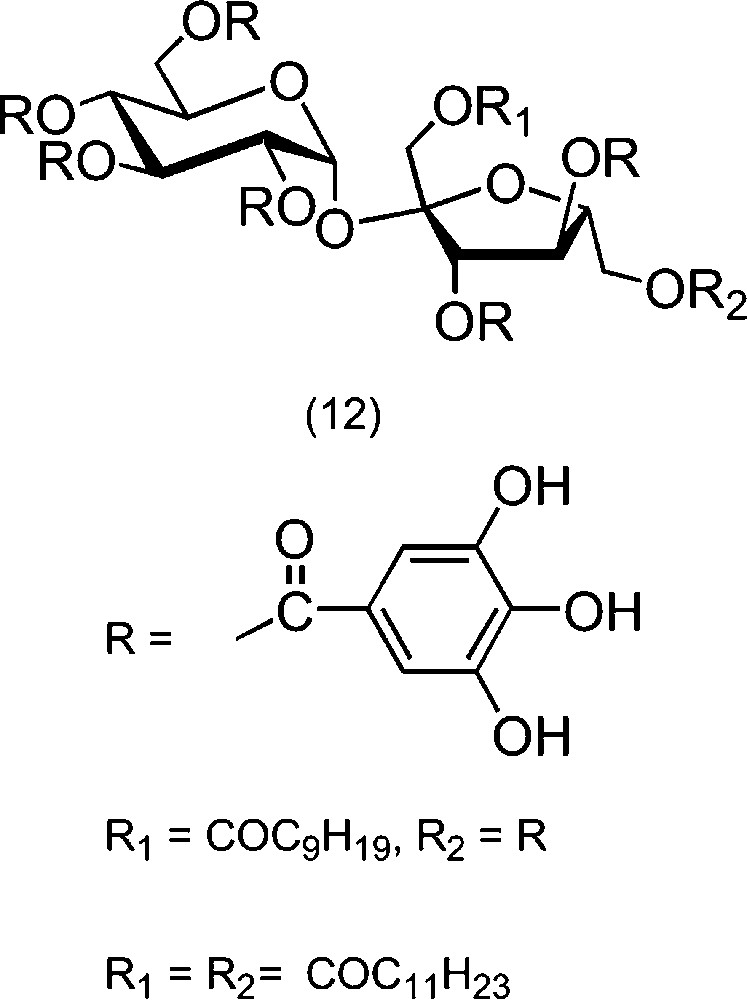
Another attractive method encompasses the use of enzymes. Hydrolases, such as proteases or lipases, have been used for the selective esterifications of carbohydrates [66,67]. These reactions have to be conducted in organic medium, in the absence of water, since they normally catalyse the hydrolysis. The difficulty is to set up solvent conditions for which the solubilities of sucrose and of the acylating agent are compatible with the stability (and the activity) of the enzyme [68,69]. For the precise case of unprotected sucrose (Scheme 6), proteases catalyse the formation of 1′-O-acylated derivatives (10) [70–72]. The solvent used in these reactions is often DMF, but systems working in DMSO were described more recently [73–76]. Notably, this was used for the synthesis of sucrose monomethacrylates, which are interesting polymerisable compounds [77–79]. Other proteases were also shown to lead to esters at OH-2 of sucrose [80,81]. Once monoesterification is achieved, the partially hydrophobicised sucrose derivative is then more soluble in solvents in which lipases are active [82–85]. This second esterification takes place at OH-6′, and various homogeneous or mixed 1′,6′-di-O-esters (11) were prepared by this method. Some esters of this kind were acylated at the remaining OH groups with galloyl residues and compared to pergalloyl sucrose ester (12) and other derivatives in the same family with respect to their antioxidant activity (Scheme 7) [86,87]. Carbamates were also prepared from sucrose and isocyanates in organic [46,88,89] or in aqueous medium [90].
4 Acetalation of sucrose
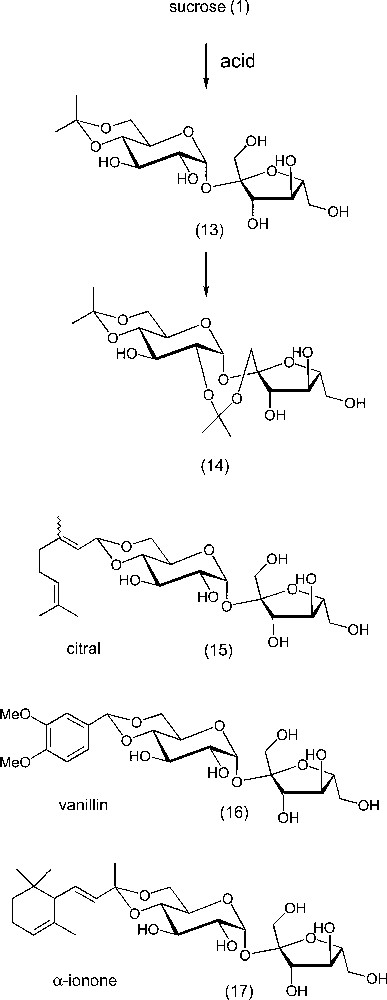
Carbohydrates react with carbonyl compounds under acidic catalysis to form cyclic acetals [91,92]. This reaction is of considerable interest for protection purposes, and is a ‘classic’ in multi-step syntheses of complex carbohydrate derivatives and oligosaccharides. In the case of sucrose, the fragility of the glycosidic bond of sucrose under acidic conditions (due to both its fructofuranosidic and bis-acetalic characters) is problematic in many cases. Only highly reactive carbonyl compounds can be used in conditions in which the disaccharidic integrity of the molecule is preserved. The reaction is selective of the OH-4 OH-6 diol, typical for glucose, leading to the acetal 13 (Scheme 8). If the reaction is continued, the second acetalation will occur at the OH-2 OH-1′ diol, forming the eight-membered ring diacetal 14 [93–96]. An alternative is to use more reactive carbonyl compounds such as α,β-unsaturated aldehydes, or better, their dialkylacetals. Ethylenic acetals prepared by this way were shown to be interesting polymerisable derivatives [97]. Among α,β-unsaturated carbonyl compounds, citral and ionones are intermediates in the fragrance industry. Sucrose acetals 15 and 16 of these compounds, and of vanillin (17), an aromatic aldehyde, were formed under mild acidic catalysis (e.g., pyridinium p-toluenesulfonate) [98,99]. The active molecules are temporarily protected, since their reactivity (notably with oxygen) is essentially due to the presence of the carbonyl group. The hydrolysis is also easy and the fragrance can be released upon slight pH change. Furthermore, the water solubility of the conjugate can be another positive aspect of the coupling with the sugar. In the cases of the more reactive carbonyl substrates, the reaction could be performed under heterogeneous acidic catalysis based on lanthanide salts [100].
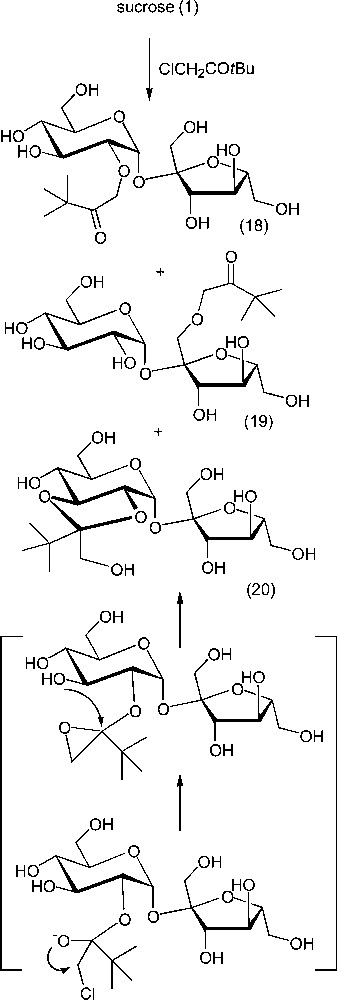
Sugar acetals can also be formed under basic conditions, from gem-dihalogeno substrates, for example [91,92]. Alternatively, reaction of sucrose with an α-chloromethyl ketone (α-chloromethyl t-butyl ketone) in the presence of a base led to a hydroxymethyl acetal involving OH-2 and OH-3 (20) as the major product (Scheme 9), besides ethers 18 and 19 arising from a Williamson reaction at OH-2 and OH-1′ [101]. Although the acetal linkage is not as stable as that of an ether, the conditions do not allow the reversibility of the process. This acetal is produced the attack of the carbonyl groups of the α-chloromethyl ketone by OH-2, leading to an hemiacetal anion that, upon cyclisation, forms a three-membered ring acetal, subsequently opened at its very reactive acetalic centre by the nearest hydroxy group (OH-3). In terms of relative kinetic reactivity in sucrose, both the acetal and the ether at OH-2 are resulting from the preeminent reactivity of OH-2, accounting, in some conditions for up to 90% of the monosubstituted products.
5 Oxidation reactions
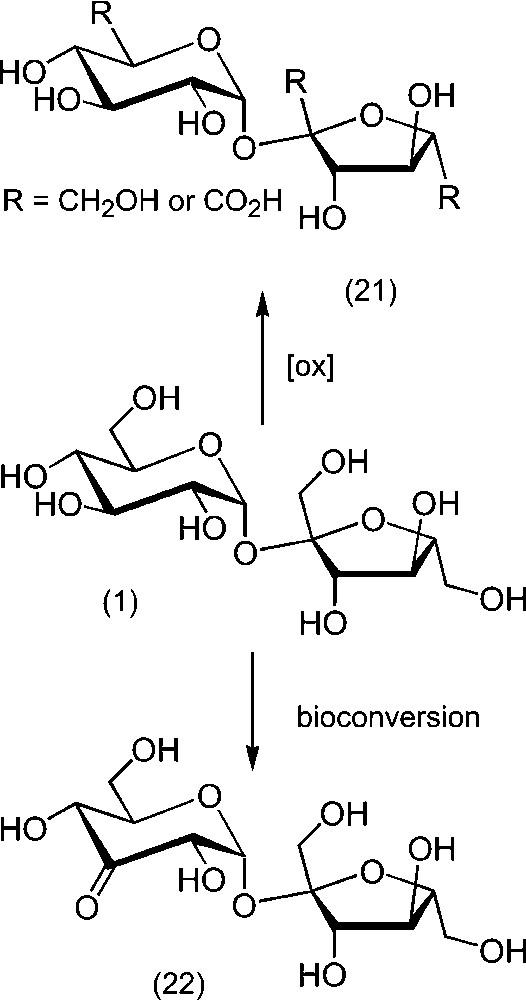
Due to their cation-sequestering properties and their potential industrial applications in detergency, some carbohydrate derivatives having carboxylic residues obtained by oxidation have attracted constant attention [102,103]. Oxidations reactions of sucrose can be divided in two types, depending on the conservation of the disaccharidic backbone or not. For example, lead tetra-acetate or periodic oxidation produces compounds in which the cyclic structure of either the glucosyl or the fructosyl moiety is cleaved, leading to interesting polycarboxylated compounds [104–108]. In contrast, oxidation by oxygen in the presence of platinum provides derivatives with carboxy groups at the primary positions [109–111]. Mono-, di-, and tricarboxy-derivatives (21) are obtained, with possible isolation of the monocarboxylated derivative by continuous extraction from the medium (Scheme 10) [112]. Electrocatalytic oxidation also led to derivatives with a carboxy group at the primary positions, although complex mixtures with more degraded compounds were obtained [113]. Concerning the secondary hydroxyl groups, oxo derivatives can be obtained via chemical or biochemical processes. For example, 3-oxo-sucrose (22) is obtained by action of the d-glucoside 3-dehydrogenase, which is the active species in Agrobacterium tumefaciens. New derivatives of sucrose were prepared from 3-oxo-sucrose by reduction (to sucrose 3-epimer allose) or reaction with nitrogen nucleophiles [114–117]. Oxidation with bromine provides the 2-keto derivative as major product, together with the 4- and 3′-oxo sucroses [118].
The TEMPO-mediated oxidizing system, now recognized as a promising oxidation reagent, has been applied to carbohydrates with success, producing carboxylated derivatives at the primary positions [119–120]. The stable radical TEMPO is oxidized to a nitrosonium ion (the actual oxidant in the system) by a stoichiometric oxidant such as sodium hypochlorite in the presence of bromide ions. With respect to environmental concerns, many variations of the method have been reported, notably heterogeneous TEMPO analogues [121–125] and bromide-free methods [126–127]. This included a sonochemical version in which the reaction could also be performed in the absence of sodium bromide, providing new elements in the search for the mechanistic role of the bromide ions [128–130].
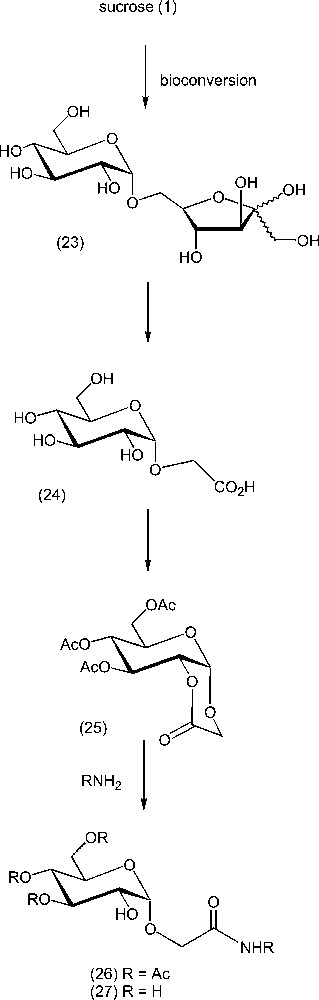
Other carbohydrates can serve as substrates for most of the processes. Among many examples, the hydrogen peroxide oxidation of isomaltulose (23), a disaccharide produced on the industrial scale from sucrose by bioconversion [131–132], gives essentially one product, namely carboxymethyl α-d-glucopyranoside (24, α-CMG), in 30% yield in an unique step (Scheme 11). Under acetylation conditions, this glucoside could be transformed to a bicyclic lactone, a most useful synthetic intermediate, as lactone opening by nucleophilic species (e.g., amines or alcohols) generates new types of carbohydrate-based materials, such as amphiphilic or polymerisable compounds 26 and 27 (after deprotection), as well as pseudo-glycopeptides when using amino acids as nucleophiles [133–134].
6 Conclusion
Among the numerous possible chemical transformations of sucrose, we have focussed on those that illustrate the links between the structure and the relative reactivity of the hydroxyl groups. By judiciously choosing the electrophilic substrate, the catalyst, or the medium, different positions of the molecule can be substituted, often with good selectivity control. Besides the classical 4,6-O-acetalation of glucosides, three main processes are observed: OH-2 selective reactions based on electronic factors, primary OH groups (mainly OH-6 and OH-6′) selective reactions based on steric hindrance and OH-1′ selective reactions based on protease catalysis. These reactions provide essentially monosubstituted derivatives showing that the regioselectivity also provides some control of the degree of substitution.
The selective processes permit direct functionalisation of the molecule, and allow its use as starting material for products of industrial relevance [135]. Many other reactions can be achieved using sucrose and are mentioned in other reviews [1–9]. Among them, the bioconversions based on transferases leading to tailored oligosaccharides are to be mentioned, since these are also green processes leading to valuable products [136–138].
Acknowledgements
The work of our group presented in this review was achieved in the former Béghin-Say–CNRS facility in Villeurbanne (CNRS UMR 143).