

1 Introduction
Molecule-based materials, in the broad sense, is a term coined essentially to make a distinction between classic inorganic atom-based solids mostly used for technological applications, such as metals, alloys or oxides, and materials built from pre-designed molecular entities. Thus, instead of high temperature synthesis typical of solid state chemistry, molecule-based materials are obtained through soft routes, traditionally from organic chemistry, coordination chemistry and supramolecular chemistry. The design of the precursors allows for a better control over the outcome of the reaction that may be even predicted. Molecules with the desired size, shape, charge, polarity, and electronic properties can be prepared, targeting for their desired role. This opens unprecedented possibilities, only limited by the imagination of the chemists, and their craftsmanship.
If molecular materials are to be competitive, they need to prove that they can mimic the properties of materials being used in current technologies, but that they can do better at a reasonable price. That is why molecular sciences are mainly flourishing technologically in those fields where they offer an added value: mostly photonics and electronics (chromophores and conjugated polymers) [1–3]. In the basic research arena, the latest and future trends where molecular materials can offer an added value point towards two different directions. Actually, molecular materials can play a crucial role controlling size and building up complexity for next generation materials.
Regarding size, as part of the nanoscience and nanotechnology revolution, molecular sciences have already many tools available to design molecules (already at the nano-scale) with the desired characteristics that can be organized at the molecular level. Therefore, although heavy concern exists on stability and reliability of “single-molecules”, it is easy to envisage that a key role could be expected for molecule-based nanomaterials in the future, once the great challenge of addressing single molecules is solved.
Regarding complexity, molecule-based materials offer really unprecedented possibilities. As in biological systems, molecule-based materials can be built from several specifically designed unique molecules, where the sum of functions could yield new or non-existing properties. In this strategy, the building blocks are prepared with the desired chemical or electronic characteristics, to be combined later into a solid state material, as a salt, crystal, polymer or thin-film, to afford the desired physical cooperative properties. Thus, in addition to the electronic properties, one needs to control the chemical character of the building blocks that will determine their intermolecular interactions and, therefore, their organization in the solid. The intermolecular interactions in play can also be predetermined, and they can go from very strong to weak, from chemical bonding, coordination bonding, hydrogen bonding and cation–anion electrostatic interactions, to host–guest, ion–dipole, dipole–dipole or Van der Waals intermolecular interactions, including any combination of them. Finally, the strength and directionality of all existing intermolecular interactions will determine the supramolecular structure of the materials, along with the interplay between the different properties.
The cation–anion electrostatic approach has been by far the most common and successful strategy, building multifunctionality by the combination of two different molecules, a cation and an anion, which have a strong tendency to self-assemble forming two-network materials with two different properties. There is essentially no limit to this approach, except for the stability of the two (or more) building blocks in the reaction media, where in the absence of any other charged species, the most probable outcome would be the combined mixed salt. The stoichiometry and crystal structure of such salt will be determined by the shape and charge of the species along the preferred supramolecular contacts in the solid.
This cation–anion approach has been explored for the preparation of different combinations of properties. Regarding dual-function materials there are several examples of combination of optical, electrical and magnetic properties that yielded optically active magnets [4], optically active conductors [5] and/or magnetic conductors [6].
This field of magnetic conductors was started in the late 1980s as part of the research on organic conductors, from the combination of cationic organic radicals able to promote electrical conductivity in self-assembled stacks in the solid state with anionic metal complexes with localized electrons responsible for the magnetic properties. As organic radicals, TTF derivatives (Fig. 1) [7,8] were the only ones really successful to yield magnetic semi-conductors, metals and superconductors [9]. On contrast, many different species of magnetic counter ions were tested from small tetrahedral or octahedral metal complexes to bulky polyoxometallates [6,10,11]. The magnetic properties were quasi-independent of the electronic properties in all cases, where the magnetic behavior of the anions remained unchanged from that of the simple salts. In the case of the tetra halides, the proximity of the paramagnetic centers promotes strong enough magnetic interactions for the appearance of antiferromagnetic ordering at very low temperatures. Although these features give rise to unprecedented phenomena, such as field induced superconductivity [12], ferromagnetic ordering was not achieved. Ferromagnetic superexchange in the anionic lattice should come into play to meet this goal.

TTF derivatives: tetrathiafulvalene or TTF (a); bis(ethylenedithio)-TTF or ET (b); bis(ethylenedioxo)-TTF or BEDO (c); bis(ethylenediseleno)-TTF or BEST (d); bis(ethylenthio)-TTF or BET (e); and bis(ethylenedithio)tetraselenafulvalene or BETS (f).
This idea brought to the game the polymeric bimetallic oxalate-based magnets [13,14]. A family of magnets, with critical temperatures between 5 and 44 K, and that were also used for the preparation of multifunctional materials, such as photoactive magnets by combination with photoactive cations [15]. In 2000 the first ferromagnetic organic metal, [ET]3[MnCr(ox)3]·CH2Cl2, was finally discovered [16] by combination of the bis(ethylendithio)tetrathiafulvalene organic radical with bimetallic oxalate-bridged ferromagnetic layers, opening the area of ferromagnetic organic metals, and the first step towards ferromagnetic organic superconductors (Fig. 2). Here we will go through the state of the art in this area, attending to all the data and details that have been gathered in recent years.
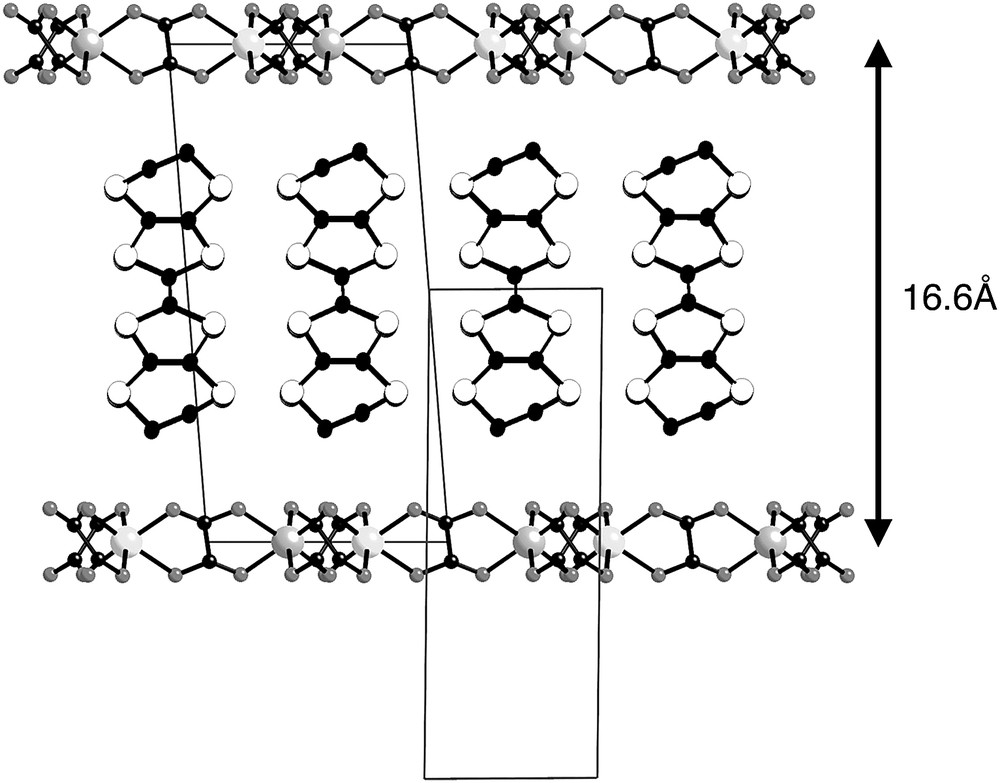
Representation of the multilayered structure of [ET]3[MnCr(ox)3]·CH2Cl2 with the two sub-unit cells.
2 Synthesis: from building blocks to solid state
Radical salts of the TTF family are usually prepared by electro-oxidation of the neutral species in a solution containing the desired anion. Once the radical cations are generated at the electrode, the corresponding salt crystallizes, with the stoichiometry controlled by the match of size and shape of the building blocks. Polymeric anions have also been used in the past, from soluble precursors [17]. The synthetic challenge appears when the anion is not only polymeric but insoluble, as happens with the bimetallic oxalate complexes. It is known that even in the absence of any other cation, a solution containing a divalent metal and a trivalent metal oxalate complex yields an insoluble oxalate-bridged 2D polymer [18].
Therefore, the key step consisted in finding a mixture of solvents where the building blocks to the inorganic network, namely a divalent metal and a [MIII(ox)3]3− complex, could be maintained in solution for long periods of time. These mixtures of solvents also had to be able to dissolve the neutral organic radical whereas, and equally important, the product had to be essentially insoluble. Any aficionado can guess that no single solvent can meet all these requirements. By a slow and long process of trial and error we could find a good enough solution by mixing methanol, dichloromethane and benzonitrile (PhCN). In this case not only the right mixture and proportions resulted in the solution to the synthetic challenge. The whole process, with attention to the order in which solvents and reagents are added, had to be optimized for success. The general procedure starts form a clear methanolic solution (20 mL) containing 1 mmol of the MII cation and the [MIII(ox)3]3− anion in a 3:2 proportion. Twenty milliliter of PhCN are added slowly to this solution, provoking the precipitation of part of the {[MII]3[MIII(ox)3]2} salt, that is again filtered out. Finally 10 mL of CH2Cl2 are added, and the solution filtered again to remove any trace of remaining solid particles. This solution, without further treatment, is placed in an electro-crystallization U-shape tube, with 10 mg of the neutral form of the TTF derivative located in the anodic compartments. Crystals of the desired hybrid material are obtained by passing a current of 0.1 μA for a few days.
Following one of the great advantages of molecular materials, this very same synthetic procedure could also be adjusted for other different building blocks to tune the physical properties towards tailor-made materials. This can be very valuable for the understanding of the correlations between properties and structure. In this case, one can modify the conducting and magnetic properties by modifying the organic radical or the metal ions. The change in organic radical is quite feasible, and most organic radicals big enough have work successfully, producing a series of [D]x[MnCr(ox)3] analogs, with different crystalline quality [19,20]. On the contrary, smaller radicals such as TTF did not yield any solid. The reason could be the worse match between charge and sizes with regards to the oxalate layer, but also the higher solubility of the final product, that could preclude the growth of a solid on the electrode.
The use of different metal ions was much more difficult, and it was only successful in a few cases. Two main considerations need to be taken into account: the redox stability of the ions under the applied voltage for the reaction, and solubility in the media in the long term, since several days are needed for the growth of the crystals. Substitution of the MnII ion by other first row metals works well only with CoII [21]. NiII is an anomalous metal ion in this sense, and even in regular salts it could not be obtained as part of the honeycomb in the past [22]. Regarding Fe and Cu, the high stability of their oxalate complexes promotes precipitation of by-products. The substitution of the trivalent metal as the tris(oxalate) complex has also proven as difficult. All attempts with the [Fe(ox)3]3− or [Ru(ox)3]3− resulted in decomposition products from the instability of these reagents after hours in solution under electro-chemical conditions. Only the very inert and diamagnetic [Rh(ox)3]3− was successfully tested. In Table 1, there is a list of the materials that were obtained until now as single crystals or poly-crystalline samples.
Known materials in the family of layered conductors [D]x[MnCr(ox)3] with their main magnetic and electrical properties
| MIII | MII | Donor | x | Organic packing | Tc (K) | σRT (S cm−1) | Conduction type | Magnetic exchange | |
| Cr | Mn | ET | ca. 3 | β | 5.5 | 250 | Metal | Ferro | Single crystals |
| BEST | ca. 2 | ? | 5.6 | <10−6 | Insulator | Ferro | Poly-crystalline | ||
| BETS | ca. 3 | α | 5.2 | 23 | Metal/semi | Ferro | Single crystals | ||
| BET | ca. 3 | ? | 5.6 | 4 | Metal/semi | Ferro | Poly-crystalline | ||
| BEDO | ca. 3 | ? | 5.2 | 2 | Metal/semi | Ferro | Poly-crystalline | ||
| Cr | Co | ET | ca. 3 | β | 9.2 | 1 | Metal/semi | Ferro | Poly-crystalline |
| BEST | ca. 2 | ? | 10.8 | <10−6 | Insulator | Ferro | Poly-crystalline | ||
| BETS | ca. 3 | α | 9.2 | 2 | Metal/semi | Ferro | Poly-crystalline | ||
| BET | ca. 3 | ? | 13.0 | 21 | Metal/semi | Ferro | Poly-crystalline | ||
| Rh | Mn | ET | 2.53 | β | – | 13 | Metal/semi | Para | Single crystals |
3 Structure: incommensurate phases
All X-ray diffraction data available indicate that most of these materials are indeed incommensurate composite crystals and, therefore, non-stoichiometric. Two unit cells mutually incommensurate co-exist in the solid corresponding to the inorganic and to the organic sublattice, respectively. One property of composite crystals is that if only the Bragg reflections of one sublattice are considered, the structure of the other sublattice appears disordered. This was observed for the ET and BEST salts, where only the organic layers could be located. The reflection data obtained for the inorganic sublattice were too weak and couldn't be analyzed. In fact, crystallographic data for the inorganic layer could be only obtained when one of the first row transition metals was substituted for the heavier RhIII ion in the inorganic honeycomb network [23]. In this case, both existing sublattices could be found. The two unit cells present in the composite crystal were found to have the same c∗ reciprocal lattice parameter within the accuracy of the experiment. This confirms that the layers do not penetrate each other, leading to the requirement that the periodicity perpendicular to the layers is identical for both unit cells. On the contrary, the periodicities parallel to the layers do not match, and appear independent to each other. The low intensity of the X-ray data made impossible to find any information regarding the relative orientation between the two sublattices.
The inorganic lattice shows the typical honeycomb hexagonal 2D lattice found in the bimetallic oxalate-bridged 2D magnets (Fig. 3a), where each metal is connected to three other metals through the oxalate bridge, and with the two metals occupying alternating positions through the network. In this case, the layers were found to appear eclipsed with respect to each other. This type of stacking is the most common when the counter cations do not penetrate into the holes of the honeycomb lattice [22]. A disordered solvent molecule occupies the hexagonal holes, which should correspond to dichloromethane, in agreement with elemental analysis and size restrictions imposed by the cavity. The inorganic lattice is identical in all hybrids, since the rigidity of the oxalate network does not allow for any deviation from the honeycomb. Its presence has been always confirmed by chemical analysis and magnetic data, when structural data were not available.

(a) Structure of the inorganic layers {[MIICrIII(ox)3]·solvent}−; (b) structure of β packing in the organic layer in [ET]3[MnCr(ox)3]·CH2Cl2; (b) structure of α packing in the organic layer in [BETS]3[MnCr(ox)3]·CH2Cl2.
The organic layer, on the other hand, allows for some variations of their structural motifs in the different compounds as one can change the organic donor. The 2D packing is controlled by the preferential self-stacking of the organic radicals, so by changing the functionalization of the organic donors one can promote different types of packing and, therefore, modify the electrical properties.
ET molecules adopt a typical β arrangement (Fig. 3b), made up by a pseudo-hexagonal arrangement of ET molecules with their mean plane parallel to each other. The closest S–S distances are side to side (about 3.5 Å) to four adjacent molecules, giving the layer a clear 2D character. Instead of appearing perpendicular to the inorganic layers, the radicals show a canting close to 45°.
When selenium atoms substitute the core sulfur atoms in the TTF skeleton of ET, the structure of the organic phase changes. In the bis(ethylendithio)tetraselenafulvalene (BETS) salts [24] the packing motif for the organic radicals follows that of a typical α-phase (Fig. 3c). In this case the organic molecules are not parallel to each other, but running in chains with two different orientations and a dihedral angle of 131°. The shortest S–S contacts are again close to 3.5 Å, but the different orientation promotes for a weaker orbital overlap. The interlayer separation is also larger in this case, because the radicals appear almost perpendicular to the inorganic layers, just side-tilted by 15° from the plane normal to the layer.
The structure and packing of the organic layer in the rest of the derivatives are still unknown due to the lack of single crystals of good enough quality for X-ray diffraction analysis. The stoichiometry found for all of them is similar to those where structural data are available, therefore it is a good hypothesis to expect similar types of packing or variations of them, and similar conducting properties. There is one exception, when selenide atoms occupy the outer positions of the sulfur atoms in ET. In this case the stoichiometry is 2:1, meaning that the bis(ethylenediseleno)tetrathiafulvalene (BEST) molecules have a nominal charge of +0.5. This suggests that the BEST molecules could appear as strongly coupled dimers.
The closest contacts between the two independent layers, organic and inorganic, appear to be from the ethylenic groups, but no directional interactions were found. No hydrogen bonding was observed. Thus, only electrostatic interactions from the overall cationic and anionic charges in each layer combine both networks, where the intralayer packing is actually dictating the final structure. This 2D character creates a mismatch in the packing, which is responsible for the incommensurate nature of these composite crystals. Only the charge density of the organic layer is also controlled by the overall charge that is fixed in the inorganic layer.
This mismatched structure creates an intrinsic disorder. Although in the organic unit cell only one TTF derivative molecule appears as crystallographically independent this does not mean that all organic radicals will be electronically identical in the crystal. Quite the contrary, since there is no periodic pattern, each organic molecule will have a different environment. Some will be close to the oxalate molecules, some to the metal centers, some to the hexagonal holes, occupied by dichloromethane molecules. This random distribution will have small effects on the organic radicals, but can clearly affect the charge distribution, and/or electronic delocalization in the conducting system.
Another crystallographic disorder is generated by the different conformation of the ethylenic groups in the organic radicals between the two possible boat conformations. This has also been discussed as important enough feature to disrupt the onset of a superconducting regime in other organic conductors [25–27]. This ethylenic disorder can be avoided by the use of a chiral ET derivative, as the S,S,S,S-tetramethyl-ET (TM-ET). The resulting material represents also the first optically active ferromagnetic metal, a true multifunctional material where three physical properties co-exist [28].
4 Dual-function materials: ferromagnetism and metal-like conductivity
Regarding the physical properties, all compounds present analogous behavior according to their fixed structural motif maintained throughout the series.
Magnetic measurements on powder samples and single crystals show that ferromagnetic ordering arises at low temperatures, from the parallel alignment of the spins in the oxalate layers, as found for the analogous salts reported for a variety of electronically “innocent” cations [10]. The critical temperatures are also identical confirming the presence of the bimetallic oxalate-bridged honeycomb network in all cases. No effect is observed regarding the presence, in this case, of a conducting layer in between the magnetic layers instead of an insulating layer as in previous cases. There is a very small difference observed in the magnetization curve (Fig. 4), where saturation is reached faster in the conducting compounds, although the longer interlayer separation could also contribute to this extent.
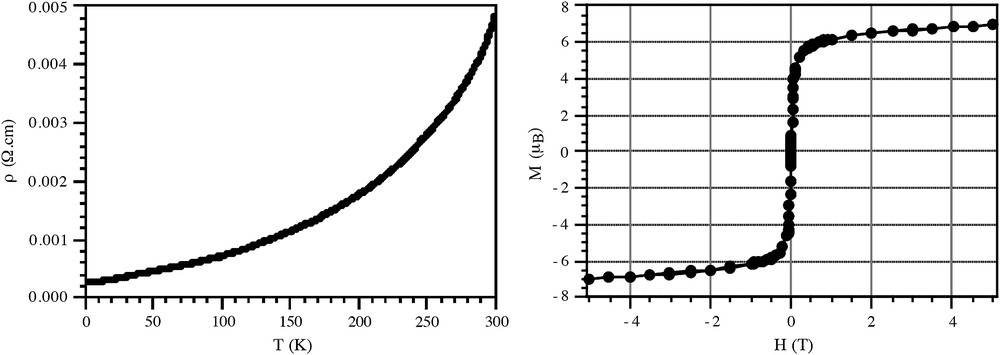
Single-crystal temperature and field dependence of the electrical conductivity (left) and magnetic hysteresis loop at 2 K (right) for [ET]3[MnCr(ox)3]·CH2Cl2.
Transport measurements on single crystals and pressed pellets indicate that all materials of formula [D]x[MnCr(ox)3] (2.5 < x < 3) behave as metals, with conductivities between 1 and 200 S cm−1 at room temperature. The ET derivative remains metallic down to 0.3 K (Fig. 4) and under pressure [29], but none of the other salts do. In all the other cases a minimum in resistance is reached below 150 K, and the resistance starts to increase down to very low temperatures. In general, this cannot be essentially regarded as a transition into a semi-conducting state, since the values of conductivity at very low temperatures are usually of the same order of magnitude of those at room temperature. This behavior is typical of charge-localization processes responsible for the rise of the resistance. Charge localization in this case could be favored by the intrinsic disorder in these composite crystals, where the radical molecules are set in many different and random environments.
It is also important to note the high anisotropy of the transport properties due to the 2D nature of the packing. The conductivity in the layer is four orders of magnitude larger than the one perpendicular to the layers, although the metallic behavior is retained in all directions.
Some unusual features are observed in the conducting properties of these ferromagnetic metals, as result of the onset of the ferromagnetic ordering. The magnetoresistance behavior of single crystals below the ordering temperature shows the appearance of a negative magnetoresistance when a small external field is applied perpendicular to the ferromagnetic layers. It reaches a minimum around 2 T and then the resistance starts to increase with the magnetic field. On the contrary, when the external magnetic field is applied parallel to the layers, the magnetoresistance is positive at low fields until it reaches a maximum around 1 T, and then starts to decrease very slowly. These changes in the sign and behavior of the magnetoresistance are only observed below Tc indicating that the conducting layer feels the presence of the internal magnetic field created by the ferromagnetic layers. In the case of the 2:1 compounds, as in [BEST]2[MnCr(ox)3], the materials behave as an insulator, with values for conductivity at room temperature as low as 10−6 S cm−1.
5 Perspectives for multilayered molecule-based ferromagnetic conductors
It is clear that the biggest challenge in the search for molecular ferromagnetic conductors, and especially for ferromagnetic superconductors, remains in the synthesis, and particularly in the design of the building blocks. The oxalate complexes show very good advantages regarding their solubility and flexibility and since ferromagnetic ordering is already achieved, they may remain the best candidates as magnetic counterpart. Keeping these building blocks, further synthetic work is clearly needed for the preparation of the [Fe(ox)3]3− analogs, where magnetic ordering would occur at much higher temperatures up to 45 K [30,31], and where attractive additional phenomena appear, such as negative magnetization [32,33] or increased hardness [34].
In order to improve the conducting properties it would be important to avoid the mismatch between the TTF network with the oxalate layer to produce a crystalline stoichiometric material. Due to the rigidity of the inorganic network, a larger TTF derivative would be probably needed for single-crystal stoichiometric phases. However, this problem does not have an easy solution, because conductivity resides on the S–S contacts that must be shorter than the sum of Van de Waals radii (3.6 Å). Other types of packing could be useful, from β to κ, that could be induced by different solvent choices. All these possibilities are under study.
On the physics ground, and until superconductivity is achieved, some important questions are still open, as the metallic conductivity that was found in the direction perpendicular to the organic layers. In this case the electrons are going through the inorganic and insulating oxalate layer apparently without the opening of a gap. No mechanism has been proposed to explain these phenomena.
Acknowledgments
The authors want to thank the ongoing support from the EU (MAGMANet NOE), MEC (Projects Consolider-Ingenio in Molecular Nanoscience (CSD2007-00010), MAT2004-3849, CTQ2005-09385, CTQ2006-27186-E and CTQ2005-25211-E) and Generalitat Valenciana (Project ACOMP07-074).