

1 Introduction
Heterogeneous catalysis is widely implemented in refining processes. The purposes of such catalysts are various: Reforming catalysts increase octane index for gasoline, hydrotreatment (HDT) catalysts reduce the amount of sulphur in final products and hydrogenation catalysts transform the olefins. Catalytic metals are deposited in different ratios on large surface alumina or alumino-silicates.
Elements present in the samples are dependant on the role of the catalyst. Pt, in association with Sn, Re or In is widely used for reforming catalysts. Mo, Ni, Co and P are associated in different ways to obtain hydrotreatment catalysts [1]. Pd, in association with Au, Ag or Ni, has been found to be efficient for hydrogenation catalysts [2,3]. Si and/or Al are present in the support. Other elements (such as S, C, V, Fe,…) can appear as contaminants through catalytic cycles.
Catalytic properties are closely correlated to the nature and concentration of elements and furthermore, the use of expensive elements in catalysts' production fostered the development of new fast and accurate methods for their determination. The determination of elements in catalysts is usually done by atomic absorption spectroscopy (AAS) in a flame (FAAS) or with electro-thermal vaporisation (ETAAS), by Inductively Coupled Plasma (ICP) with atomic emission spectrometry (ICP–AES), or by X-ray Fluorescence (XRF) [4].
Since the introduction of inductively coupled plasma (ICP) techniques, trace analysis of various materials has shown great progress. Several potential advantages include low detection limits (ppm to ppb levels), high sensitivity, large dynamic range and reduced matrix effects due to the high temperature of the argon plasma [4]. The use of atomic absorption spectrometry (AAS) also gives high performance for the determination of trace elements in catalysts [4,5]. The limitation of the latter technique is the determination of only one element per analysis. Generally, prior to analysis, solid samples have to be digested with concentrated acids [4–7]. The sample preparation step can introduce contaminations through addition of acids, this step is time consuming and some difficulties may arise from sample digestion. At the end of the sample preparation, the final solution is homogeneous and not affected by initial sample heterogeneities.
The direct determination of elements in solid catalysts can be done by X-ray Fluorescence. For truly quantitative analysis, attention must be paid to the heterogeneity of the sample and the smoothness of the surface, because the characteristic X-rays escape from the first few μm of the sample [6]. Truly quantitative analysis can only be obtained if a large number of grains with homogeneous composition are sampled. A simple grinding procedure (below 100 μm) is necessary to ensure a homogeneous analysis of minor elements. This method, often employed, has the advantages of simple sample preparation and rapid measurement for the analysis of minor elements in samples like auto catalysts [8] or monometallic catalysts [4]. A longer preparation step, including addition of lithium tetraborate, is employed for the accurate determination of major elements (Si, Al or Ni, Co, Mo) in hydrotreatment catalysts. This sample preparation, as for acid digestion, can introduce limitations related to time consumption (several hours) or sample contamination. All elements with atomic number higher than 11 (Na), can theoretically be determined.
A combination of solid sampling using laser ablation hyphenated to inductively coupled plasma (LA–ICP) techniques developed in view of avoiding digestion step and to benefit from multielemental capacity and high sensitivity of the ICP–atomic emission spectrometry (LA–ICP–AES) or ICP–mass spectrometry (LA–ICP–MS) [9–11]. The impact of a laser beam on the sample surface is employed to sample the solid material and produce particles transported into the ICP. This method has already been widely used for the direct analysis of many samples, especially in geology, metal analysis or glass analysis. The main advantages are: reduction of the time for sample preparation and limitation of contamination risks. For laser ablation, like any solid sample analysis method, heterogeneities in sample composition will lead to false and variable results for the bulk analysis of the sample. The direct analysis of a solid material by LA–ICP may be achieved after grinding of the material and deposition of the powder on a sticky support, but the signals obtained are very unstable [12]. To ensure a homogeneous sampling, and further a stable signal by laser ablation, the powdered materials are generally pressed into pellets in order to obtain a flat and homogeneous target surface [13,14].
Although LA–ICP (AES or MS) was introduced in the mid 1980s [15], applications to the characterisation of catalysts are rare [16–19]. Baiker et al. determined titanium and platinum group elements (PGEs) in flame catalysts. For Ti, samples with 10–20 nm particle sizes and metal loading of 0.3–3.2%w/w Ti/SiO2 were analysed and tested for epoxidation of cyclohexanol. For platinum, the catalysts were also nanoparticles and the metal loading was higher (1–10%w/w Pt/Al2O3). These samples were evaluated for the hydrogenation of ethylpyruvate. Finally, Pt, Rh and Ru were determined in flame-made catalysts with metal content between 0.1 and 5%w/w. Authors show that Ti can be detected down to 1–40 ppm and that the determination of platinum group elements gives about 10% bias on the determination of the concentrations (a Pt–Rh–Ru catalyst, prepared on the same way and characterised by XRF, was used as standard). In a previous work [20], the analysis of Pt, Sn and In on refining catalysts was evaluated. A comparison of the standard addition method and external calibration was performed and accuracy was found to be better using external calibration (Pt 3%, Sn 11% and In 29%).
The aim of this work is to demonstrate the possibilities of LA–ICP–AES for the elemental characterisation of different catalysts. Analytical performance like dynamic range, number of analysable elements and detection limits is presented through the examples of hydrotreatment, reforming and hydrogenation type catalysts. The performance obtained is in good agreement with the objectives of such characterisations: fast, multielemental, sensitive and accurate. A detailed discussion of factors influencing accuracy is presented,insisting on the influence of the nature of the catalyst support and the physico-chemical form of the metal.
2 Experimental
2.1 Samples
For the evaluation of analytical performance, catalysts were supplied from IFP. The catalysts' support was alumino-silicates for NiW(P) samples and alumina for the others. The active phases were Ni monometallic (3 samples), Pd monometallic (3 samples), Pd–Au and Pd–Ag (1 sample for each) and Pd–Ni (3 samples) for hydrogenation type catalysts. Pt and Pt–Sn were the active phases for reforming type catalysts (30 samples). The active phase of HDT type catalysts was CoMo (P) (6 samples), NiMo (P) (3 samples) and NiW(P) (12 samples).
For the investigation of matrix effects, Pt–Sn catalysts were prepared on different alumina and with different chemical forms. Catalysts were prepared by dry impregnation of metallic precursors on ground alumina. The concentrations of precursors (H2PtCl6 and SnCl2) in the spike solutions were chosen to obtain final concentrations consistent with reforming catalysts (<1.3%w/w Pt and <0.7%w/w Sn). Hydrochloric acid was added to the spike solutions to ensure a similar amount of chlorine in all samples (1.5%w/w), necessary for a homogeneous distribution of metals throughout alumina. After the impregnation step, samples were dried at 120 °C for 120 min, and finally calcinated under air atmosphere at 560 °C for 120 min (ramp 5 °C/min). This procedure ensures the formation of particles of platinum and tin oxides on the catalysts.
2.2 Pellet preparation
Samples have been received as extrudates or 1 mm beads, with a large size distribution. Catalysts were ground to obtain a final grain size below 100 μm. This step leads to better reproducibility of the results during laser ablation analysis [20]. One gram of each catalyst was collected for analysis by wavelength dispersive X-ray fluorescence (WDXRF).
For LA–ICP–AES analysis, 400 mg of each catalyst was pressed as 13 mm diameter pellets (750 MPa pressure using 8 min pressure time, hydraulic press type LP25 from Ligthpath optical-UK, 13 mm pellet die from Specac). Depending on the amount available, between 2 and 5 replicates were prepared for each pellet.
2.3 Instrumental parameters
The laser ablation system is the Lina Spark™ atomizer (LSA Sarl, Cully, Switzerland), which is especially dedicated for bulk analysis. The laser beam of a Continuum Surelite Nd:YAG laser working in the Q switch mode at its fundamental wavelength of 1064 nm is moved circularly over the sample surface by means of a motor driven lens. The resulting diameter of the ablated area is roughly 3 mm. The laser beam is focused within the target giving a spot diameter of roughly 1 mm. Optimisation of the defocalisation of the IR laser beam was previously studied in our laboratory and was demonstrated to be especially appropriate for bulk analysis [21]. In this work a repetition rate of 10 Hz was employed and a fluence of 7 J/cm2 was selected for an optimum ablation rate on the pressed powders. With these conditions, ablation depth resulted in about 0.1 μm ablated per shot for all samples. The pre-shot delay of 150 seconds, necessary for signal stabilisation, results in the removal of 150 μm. The acquisition time of 30 seconds results in an analysed depth of nearly 30 μm. Laser ablation sampling is therefore involving the bulk composition of analysed catalysts.
The ICP–AES is a Varian Vista-Pro (Varian – Australia) equipped with a 40 MHz generator and an echelle grating based dispersive system. The viewing is axial, and the CCD detection allows monitoring simultaneously the analyte lines. Laser produced particle aerosol is mixed with a nebulized aqueous solution using a Scott type spray chamber specially modified to allow introduction of the two aerosols. A detailed description of the hyphenation is presented in Ref. [20].
3 Results and discussion
3.1 Laser ablation–ICP for catalysts' characterisation: sample preparation and figures of merit
Sample preparation for laser ablation is necessary to obtain flat and homogeneous surface, the preparation step is fast and improves signal stability.
3.1.1 Sample preparation
In previous work pellets were prepared with 1 g of catalyst. In order to reduce catalyst consumption, 13 mm pellets are prepared. The pelletizing conditions used for ground catalysts in this study were optimized and are presented in experimental part.
Mechanical stability of pellets prepared from Al2O3 or alumino-silicates is satisfying for LA–ICP analysis. In our study special care had to be taken for corundum based catalysts or alumino-silicates containing more than 70%w/w SiO2. Pellets are solid enough to resist the ablation process, but can easily be broken during sample handling. The analysis of 5 pellets prepared from the same ground sample showed that sample preparation generated less than 5% variation.
Sample preparation in LA–ICP is quicker than digestion or fused pellet preparation. Furthermore, the pellet being pressed without any binder, the only risk of contamination comes from the pellet die and the cylinders.
3.1.2 Elements of interest
In the field of catalysis, it is essential to obtain complete elemental characterisation in one step. This characterisation can include the determination of the support elements (Al, Si or group I and II elements from the support synthesis), the major elements of the active phase (transition metals), the minor and trace elements (rare earth elements, PGE and other precious metals), catalytic poisons (As, S,…) and contaminants of spent catalysts (Fe).
Atomic absorption methods used so far for the analysis of catalysts (ETAAS or FAAS) are single-element methods so that a complete qualitative analysis would take hours [6]. On the other hand ICP–AES and XRF based methods can provide the simultaneous determination of more than 70 elements [4].
In our study, we have investigated the possible limitation due to spectral interferences in LA–ICP–AES. These interferences happen when an emission line of the analyte is superimposed with an emission line of one element from the matrix. The occurrence of interferences is a function of the element to be analysed and of the other elements in the catalyst. For elements exhibiting a rich spectra, (like Co, Ni, Mo, W with hundreds of sensitive lines), it is generally easy to find an emission line without interference, with a reduced loss of sensitivity (LODs remain in the ppm range).
For elements like phosphorus, whose spectrum consists of a limited number of sensitive wavelengths, interferences are more likely to be problematic. As it can be seen on Table 1, the most intense emission lines of P can be interfered by the elements from the support (Ti), from other catalytic elements (Ni, Mo, W), or from potential contaminations (Cu, Fe). Because of these interferences, less intense lines must be used, and the limits of detection can be doubled.
Potential spectral interferences for phosphorus in catalysts.
| Line (nm) | Relative intensity | Interferences (nm) | Limits of detection (in micrograms per gram of catalyst) |
| P 177.434 | 436 | Cu 177.421, Ni 177.334 | 31 |
| P 178.222 | 285 | 77 | |
| P 213.618 | 1550 | Cu 213.598, Mo 213.606 | 36 |
| P 214.914 | 504 | Cu 214.898 | 81 |
| P 253.561 | 317 | Fe 253.561, Ti 253.588, W 253.600 | 82 |
3.1.3 Limits of detection
For determination of trace and minor elements, the detection limits generally govern the selection of the technique. This point is important for expensive elements like platinum group elements (Pt, Pd, Rh, Ru) at low concentrations or for the determination of impurities from the support (Na, Mg,…) or contaminations during catalysts lifecycle (V, S,…).
For the analysis of solutions, ICP–AES and AAS can reach very low limits of detection at ppb level for Pd, Pt [4,6,21]. However, the digestion and dilution steps reduce this performance for the analysis of solids. Typical limits of detection in catalysts are generally below 10 ppm by ICP–AES for all elements [4].
When XRF is employed the detection limits are highly matrix-dependent. The best results are obtained for the determination of heavy elements in matrixes of light elements (Pt/Al2O3) for which LODs can vary from 5 to100 ppm [4,17]. For determination of lighter elements, such as Na, the LODs are decreased down to 40 ppm for direct analysis in alumina, and can reach 0.6%w/w when analysing pellets prepared by fusion, because of sample dilutions [4].
We have evaluated the limits of detection of several elements and compared them to the values reported in the literature for other techniques: FAAS [4], ETAAS [21,5], XRF [4,17] and ICP–AES with classical solution introduction [4,6,22]. Results are presented in Table 2.
Typical limits of detection (in micrograms per gram) for elemental analysis of catalysts.
| Element | Pd | Pt | Na | Mg | Ni | Mo | Cu | Notes |
| AAS (b) | 0.02 | (2–10) | (0.5) | (0.2) | 0.01 | 0.04 | 0.02 | [4,5,7] |
| AAS (a) | 50 | 25 | 0.2 | [4,5] | ||||
| XRF | 10–100a | 5a | 400a | [4,17] | ||||
| (0.1–10)b [6] | ||||||||
| <10a [4] | ||||||||
| ICP–AES | 0.01–0.05 b | 0.03–0.08b | 0.09 | [5,7] | ||||
| LA–ICP–AES | 5 | 10 | 30 | 30 |
a Measured in the catalysts.
b Measured in solution.
The LODs for LA–ICP–AES were determined using IUPAC recommended expression: LOD = (3σ)/S where σ is the standard deviation calculated from 10 replicates of the background measurement and S is the sensitivity (i.e. net signal divided by concentration). For every element, the limit of detection by LA–ICP–AES was below 50 ppm.
For the determination of Si and Al in the supports, these LODs are more than acceptable for those elements with concentrations varying from a few percents to tens of percents. For catalysts with high metal content, such as HDT type catalysts, the method is sensitive enough (LODs of 30 ppm) to analyse elements such as Co, Ni, Mo and P at percent level. Finally, for Pt and Pd, between 0.05 and 1%w/w in the catalysts, LODs of 5 and 10 ppm show that the LA–ICP–AES is also suitable for catalytic elements at very low concentrations.
3.1.4 Repeatability
Repeatability is important for the interval of confidence on the final results. If the method suffers from poor repeatability, analytical results will be given with high incertitude. Two repeatabilities have to be distinguished. First, the repeatability of the measure, determined by successive replicates of one measure. Second, the whole process repeatability that takes into account the sample preparation. This criterion was studied for elements at concentrations varying from 0.1 to 1%w/w for the minor elements (Pd, Pt, Ag, Au, Sn) up to several %w/w for the major elements (Co, Ni, W, Mo, Al, Si). Repeatability of the measure by LA–ICP–AES was below 5% for all elements. The reproducibility of the method was also below 5%, but could reach 10% for analytes below 0.5%w/w.
From these points, we can conclude that LA–ICP–AES analysis has repeatability similar to other techniques (generally 0.5–3% for ICP and XRF [4]) and that samples' heterogeneities do not affect the analysis.
3.1.5 Dynamic range
Generally, concentrations of precious metals like Pd or Pt are below 0.5%w/w in reforming and hydrogenation catalysts. According to their lower activity, other metals (such as Co, Ni,…) are usually more concentrated and range from 1 to more than 15%w/w. To cover a large range of concentrations and evaluate the linear range, two calibration sets were prepared containing from 0.024 to 1.1%w/w Pt and from 1.5 to 15%w/w Ni.
Net signals normalized to aluminium (internal standard) as a function of sample concentrations (determined by XRF) are displayed for platinum (Fig. 1) and for nickel (Fig. 2). It can be seen that the calibration curve, is quite linear. This linearity is observed at low and high concentrations and confirmed by the correlation coefficients varying from 0.97 to 0.999. The uncertainty at the centroid is about 10%. These values are similar to those from previous studies on the evaluation of the linearity of LA–ICP–AES [23,24]. Furthermore, the linearity is observed on more than one order of magnitude. For platinum, the response remains linear between 0.024 and 1.1%w/w, which is 45 times more concentrated. For Ni and Mo (data not shown, but results are similar to Ni), it is shown that the response remains linear from 1.5%w/w to very high concentrations up to 15%w/w. This allows the determinations of trace, minor and major elements, with only one calibration curve.
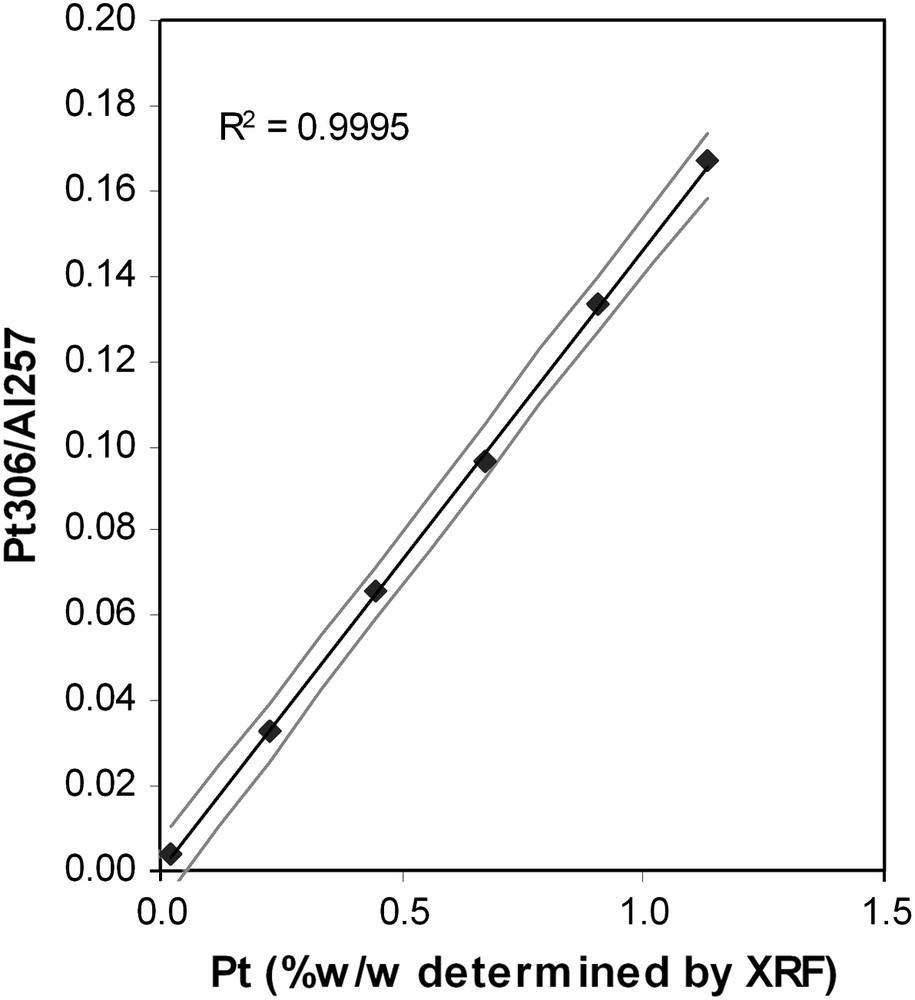
Calibration curve for Pt 306 nm, internal standard Al 257 nm. (Grey lines correspond to uncertainty of 5% on the slope of the calibration curve). Calibration set prepared by dry impregnation of H2PtCl6.
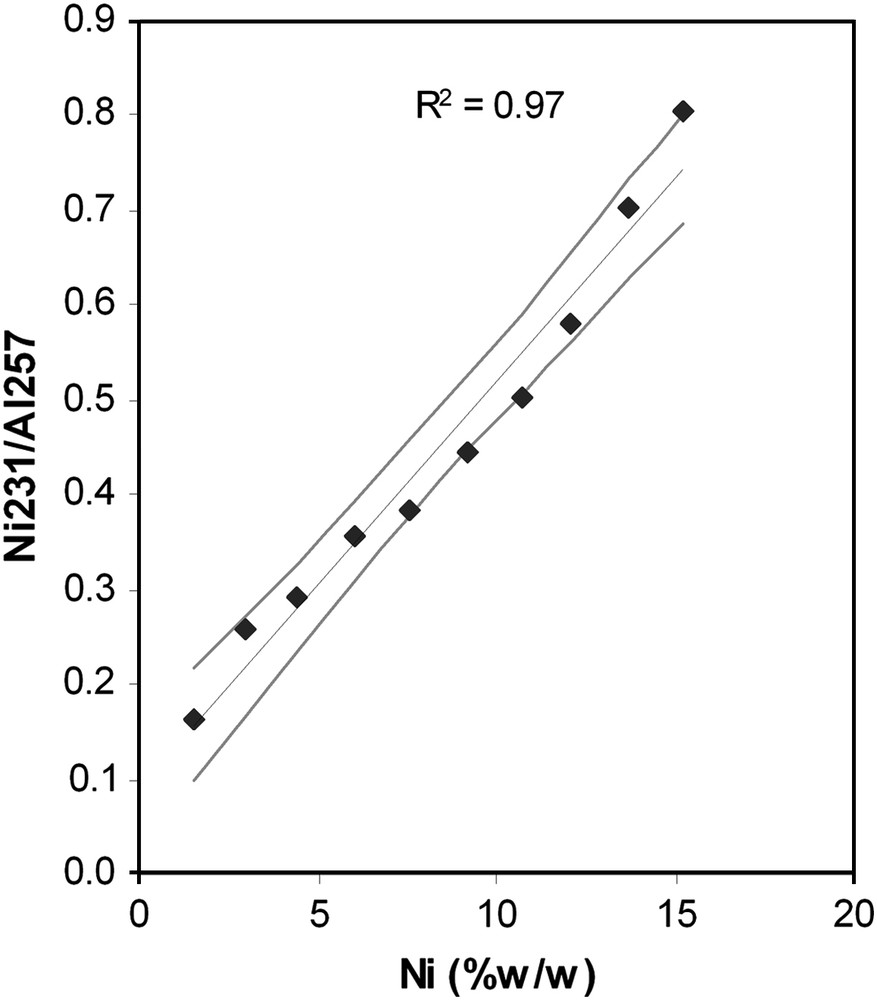
Calibration curve for Ni 230 nm (Grey lines correspond to uncertainty of 5% on the slope of the calibration curve). Calibration set prepared by solid dilution of NiNO3/Al2O3 into γ-Al2O3.
3.2 Matrix effects and accuracy
A matrix effect is observed when a slight variation of the sample matrix produces a variation of the sensitivity for a given element. Such effects are present in any analytical technique and are usually compensated or corrected using specific solutions. A good accuracy may be obtained only if matrix effects are minimized or/and compensated. In the evolution of atomic spectroscopy, the trend is to reduce such effects. As an example, when moving from flame atomic absorption spectroscopy to inductively coupled plasma, as the temperature of the second is 3–4 times higher, the so-called chemical effects are drastically reduced. Catalysts are prepared using different supports and may undergo various treatments. When solution is analysed using ICP–AES, the matrix effects are compensated using an internal standard. A standard addition method can also be employed.
In solid sample analysis, when an internal standard is not available or possibly added, the accuracy of the results relies on the availability of matrix matched solid standards (very limited for catalysts). The use of internal standards, like X-rays generated from the source in XRF, can significantly compensate for some matrix effects such as the influence of S on Ni and V [4]. Matrix effect in XRF can also be corrected with the rigorous application of matrix correction procedures [6,25]. These procedures require accurate knowledge of the fundamental parameters involved in X-rays generation, and of the sample composition.
In LA–ICP, matrix effects generally occur during the ablation, the transport or into the ICP. Ablation-related matrix effects are controlled by thermal and optical properties of the samples. ICP-related matrix effects are resulting from the inefficiency of energy transfers from the plasma to the sample. The differences in ablation are generally produced by differences in the matrix. These differences in the case of catalytic elements may include: nature of the support (i.e. phase of the alumina in our examples), physico-chemical form of the supported metals (i.e. oxide, chloride,…) and concomitant elements (i.e. Ni present together with Pd or W,…). The use of an internal standard is generally efficient to compensate for the differences in the ablated mass.
3.2.1 Nature of alumina crystalline phase
For element characterisation in alumina-based catalysts, it is important to investigate the influence of the alumina crystalline phase. For this study, the example of Pt–Sn supported catalysts was selected. The crystalline phase of the matrix could influence the analysis because of the differences in specific surfaces (from few m2/g for α-Al2O3 to 500 m2/g for η-Al2O3) or thermal behaviours. Under high temperature treatment, η-Al2O3 and γ-Al2O3 are decomposed to α-Al2O3, which is the thermally stable state of alumina. The thermal treatment of γ-Al2O3 will successively give δ-Al2O3, θ-Al2O3 and finally α-Al2O3. This transformation also modifies the porosity of the different alumina [26].
Pt–Sn catalysts, with concentration range from 0.05 to 1.2%w/w for Pt and from 0.05 to 0.06%w/w, were prepared on η-Al2O3, γ-Al2O3 and α-Al2O3. Catalysts were analysed by LA–ICP–AES and the net signal for tin and platinum (with Al as internal standard) was plotted as function of the concentrations measured by XRF. To evaluate the influence of alumina phase, the slopes (b1) and their uncertainty (tSb1 calculated on a 95% confidence level) are compared in Fig. 3.
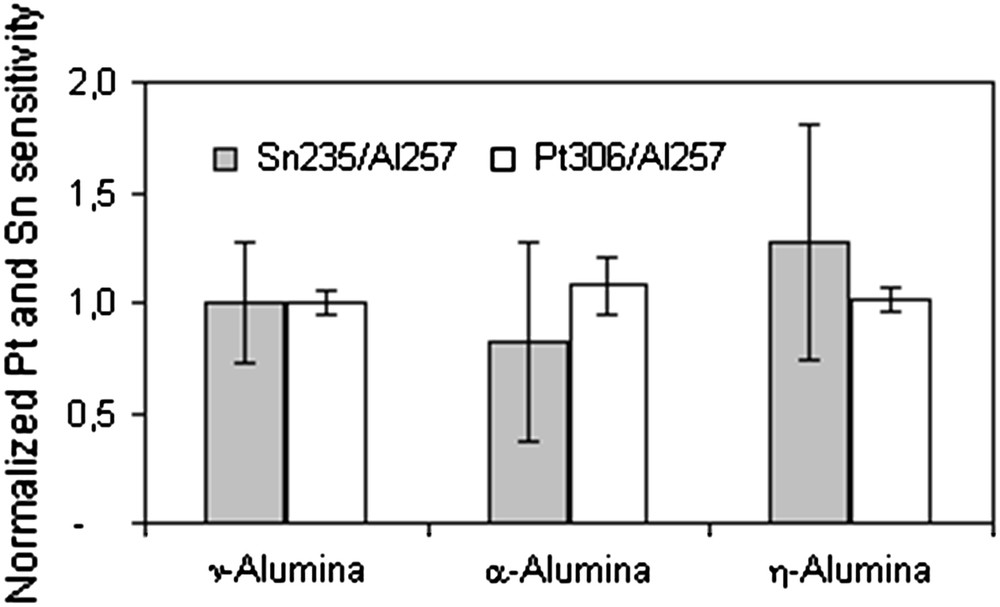
Influence of the alumina crystalline phase on the sensitivity (Pt 306 nm/Al 257 nm and Sn 235 nm/Al 257 nm). Uncertainties (95%) of the slopes of calibration curves are presented for five standards measurements.
The platinum response is very linear, with correlation coefficients over 0.99. Slopes of calibration curves are close from one alumina to another and, considering the uncertainty, the slope is similar.
Linearity of the calibration curves is less satisfactory for tin with correlation coefficients between 0.96 and 0.990. Nevertheless, the comparison of the slopes and uncertainty shows that the analysis of tin on tested alumina is similar.
Considering the overlapping of the slopes of calibration curves, the analysis of tin and platinum is similar on γ-Al2O3, η-Al2O3 and α-Al2O3. It can be concluded that the structure of the support does not influence the LA–ICP–AES analysis of catalysts and, thereafter, that catalysts prepared on different alumina phases can be analysed by the same calibration curve.
3.2.2 Physico-chemical form of the supported metal: example of platinum and tin
The influence of the physico-chemical form of the metals was studied for platinum and tin on γ-Al2O3. Previous studies pointed out that the chemical form of the elements could lead to strong matrix effects. Motelica-Heino et al. studied the analysis of synthetic geological powders (CaCO3 and SiO2) spiked with different crystalline compounds of Mg, Al and Fe (phosphates, oxides, sulphates). Results showed that the ablation efficiency depends significantly on the chemical form of the element. These differences were attributed to the different melting temperatures of the compounds (the lowest ablation rate is obtained for thermally stable forms like oxides) [14]. For the analysis of polymers, the chemical forms of the additives (Ti, Sn, Ca) also induced modifications in the sensitivity of the elements in LA–ICP–AES. It was shown that inorganic forms of these elements (Ca(OH)2, TiO2) were more refractory to ablation than organometallic forms [27]. For the analysis of catalysts, this phenomenon can be a strong limitation for samples prepared from different formulations or for the analysis of samples at different steps of catalysts' manufacturing.
For this study, we compared the response of platinum and tin as precursors (H2PtCl6 and SnCl2) and oxides. Pt–Sn catalysts prepared on γ-Al2O3, with and without a calcination step at 500 °C, were analysed by LA–ICP–AES and the signal of tin and platinum (with internal standard) was plotted as function of the concentrations. To evaluate the influence of the chemical form, the slopes (b1) and their uncertainty (tSb1 calculated on a 95% confidence level) were compared.
The results, presented on Table 3, clearly show that the sensitivity is similar for precursors (H2PtCl6 and SnCl2) and oxides for platinum and tin. It can be concluded that, for Pt–Sn catalysts, there is no matrix effect related to the chemical form of the element. In-process control of the metal content during the sample preparation can be considered with only one calibration curve.
Characteristics of the calibration curves for Pt and Sn as precursors (H2PtCl6 and SnCl2) or oxides (after 2 h calcinations at 500 °C).
| Pt 306 nm/Al 257.509 nm | Sn 235 nm/Al 257.509 nm | |||||
| b1 | tSb1 | r2 | b1 | tSb1 | r2 | |
| Oxide | 0.15 | 0.01 | 0.998 | 0.011 | 0.003 | 0.98 |
| Precursor (Cl) | 0.13 | 0.02 | 0.997 | 0.012 | 0.002 | 0.98 |
3.2.3 Physico-chemical form and concomitant metals: example of nickel
To further illustrate the accuracy likely to be obtained using LA–ICP–AES, evaluation was made on Ni containing catalysts. Two catalysts are Ni monometallic (10–20%w/w), three are PdNi (below 1.5%w/w), three are NiMo (1–5%w/w of Ni) and ten are NiW(P) catalysts (1–10%w/w of Ni). NiW(P) samples are supported on alumino-silicates, whereas the other catalysts are supported on alumina. Thus internal standardisation was corrected using the true aluminium concentration in every sample (measured by XRF).
Sensitivity for Ni determination is compared between the samples of a group and between the groups of catalysts. The comparison presented in Fig. 4 shows that the ten NiW(P) samples have similar sensitivity. The sensitivity is also similar for the three NiMo catalysts but slightly higher than the one observed for NiW(P) catalysts. For Ni catalysts, the sensitivity is similar to the one observed for NiW(P). For NiPd catalysts, results show that the three samples from that group have very different sensitivities, significantly higher than the one observed for the other samples.
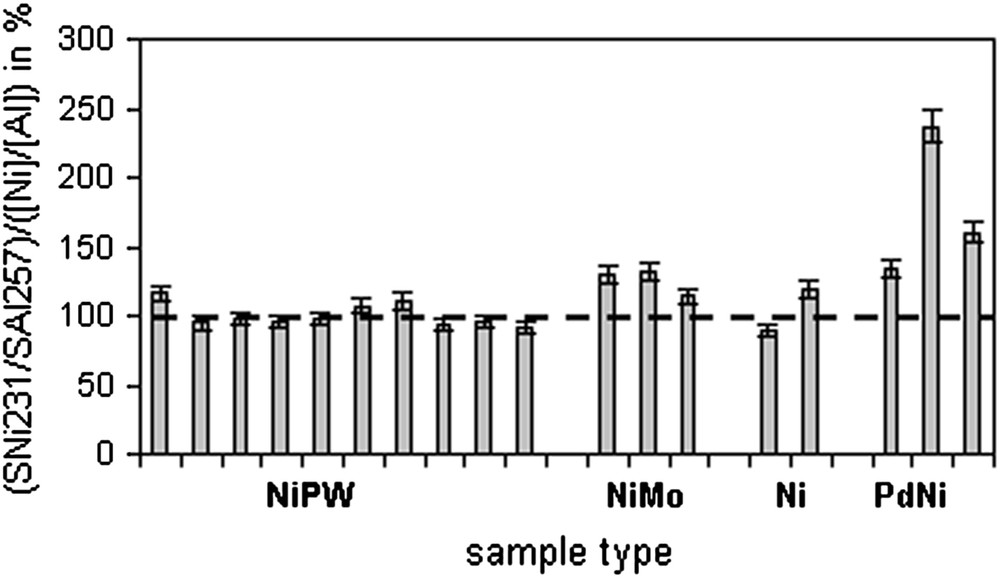
Sensitivity of Ni 231 nm with internal standardisation with Al 257 nm in 4 different matrices.
Among the 10 NiW(P) catalysts, five were selected as standards. The calibration curve obtained was used to determine the Ni concentrations of the 5 remaining NiW(P) catalysts. Other Ni containing catalysts were also analysed: 3 NiMo catalysts, 2 Ni monometallic catalysts and 3 PdNi catalysts. The bias is calculated as 100 × (Ni(LA–ICP–AES) − Ni(XRF))/Ni(XRF), where Ni(LA–ICP–AES) is the determined concentration and Ni(XRF) is the concentration measured by XRF. The results are presented in Table 4.
Determination of nickel concentrations on real catalysts with and without matrix matching (standard used: NiW catalysts).
| Ni(XRF), %w/w | Ni(LA–ICP–AES), %w/w | Relative bias, % | |
| NiW8 | 2.52 | 2.68 | 6 |
| NiW9 | 2.53 | 2.79 | 10 |
| NiW10 | 2.64 | 2.44 | −7 |
| NiW11 | 2.59 | 2.46 | −5 |
| NiW12 | 2.56 | 2.30 | −10 |
| NiMo1 | 3.35 | 4.27 | 28 |
| NiMo2 | 2.30 | 2.85 | 24 |
| NiMo3 | 3.38 | 3.76 | 11 |
| Ni1 | 19.00 | 18.31 | −4 |
| Ni3 | 7.70 | 9.58 | 24 |
| PdNi2 | 1.32 | 1.45 | 10 |
| PdNi3 | 0.32 | 0.34 | 7 |
| PdNi5 | 0.73 | 0.80 | 9 |
The differences between the concentrations obtained by LA–ICP–AES and XRF (analytical bias) were below 10% for the 5 NiW(P) catalysts. When calculating Ni in other samples (NiMo, Ni and NiPd), the bias was higher, ranging from 5 to 30%. For Ni and NiMo samples, biases were also ranging from 4 to 30%. Smaller biases (7–10%) were obtained for PdNi. This good accuracy is explained because the calibration curve better extrapolates sensitivity in the low concentrations than a simple (Signal/Concentration) calculation.
Furthermore, from this experiment it can be seen that the bias could be reduced by selecting standards similar to analysed samples (matrix matching). For instance, a NiMo calibration curve for the analysis of NiMo catalysts would decrease bias values below 10% as sensitivity is very similar (Fig. 4). A calibration curve with concentrations close to the expected concentration range in samples gives also best results.
4 Conclusion
LA–ICP–AES is a well-known method for direct determination of major, minor and trace elements in solids. Its application for the analysis of heterogeneous catalysts was very little investigated in literature.
In this work we have demonstrated that this method could be applied to the determination of catalytic metals in reforming, hydrogenation and HDT type catalysts. Compared to usual analytical methods for elemental analysis of catalysts, analytical performance is similar in terms of repeatability. The sensitivity of the method is high enough to determine simultaneously major, minor and trace (over 50 ppm) elements. The major advantage of LA–ICP–AES is the direct analysis of the sample. With sample preparation of 15 min the method is less time consuming than ICP or AAS, where the digestion step takes hours.
The Lina Spark™ atomiser associated to an ICP–AES was found appropriate for bulk analysis of catalysts as the laser spot diameter (1 mm) is larger than the usual size of active phase particles. The crater depth was found to be 150 μm, and here again, the dimension is large compared to supported metal particles ranging from few nanometres for highly dispersed metals to microns in most of the cases. As it is expected, laser ablation gives information about the bulk composition of the sample, contrary to methods like XPS or SIMS that give information about the very first nanometres of the surface.
Matrix effects were investigated to determine sources of bias on the analysis of catalysts by LA–ICP–AES. Some of the so-called matrix effects are inducing a variation in the ablated mass from one sample to the other, others are leading to a different sensitivity in ICP–AES detection. These effects are usually efficiently compensated by the classical use of an internal standard. In our study, aluminium was efficiently compensating for some of the matrix effects and investigation of accuracy led to promising results. For Pt–Sn catalysts, with low metallic charge, it was shown that the lumina phase or chemical form of the element did not influence the accuracy. For Ni catalysts, with many complex matrixes and forms, matrix effects were observed on the sensitivity. Nevertheless, the biases on the concentration were ranging from 4 to 30%, with a mean bias of 15%. When using a matrix matching between samples and standards, the bias could be reduced down to 10%. Consequently, in general case, the use of appropriate standards is important to improve accuracy.
Acknowledgement
Thanks are due to E. Roche, L. Burte, N. Texier and F. Lovery for WDXRF measurements. This work was supported by a grant from CIFRE and IFP. The authors also wish to thank the Varian Company for the loan of the Vista ICP–AES instrument.