

1 Introduction
1.1 Origine du césium radioactif
Le césium est un des produits de fission les plus abondants générés lors de la fission du combustible nucléaire (UO2 enrichi en 235U) au sein des réacteurs au cours de la production d'électricité. Ainsi, dans les solutions de produits de fission récupérées après retraitement du combustible usagé (procédé Purex) utilisé dans les réacteurs à eau pressurisée (REP), 3,6 kg de césium sont produits par tonne d'uranium pour un combustible d'oxyde d'uranium usagé enrichi initialement à 3,5% en 235U [1]. Au sein des solutions de déchets nucléaires, le césium est présent sous la forme de plusieurs isotopes radioactifs ou non (les durées de vie sont indiquées entre parenthèses) : 133Cs (stable), 134Cs (2 ans), 135Cs (2,3.106 ans), 137Cs (30 ans). Etant donnée la faible durée de vie de 134Cs, seuls sont à considérer les isotopes radioactifs 137Cs (radionucléide β,γ) et 135Cs (radionucléide β) dans le cadre de la gestion du césium. Ainsi quelques années après déchargement du combustible usagé, la composition isotopique du césium est la suivante : 133Cs (45%), 137Cs (43%), 135Cs (12%). Lors de leur décroissance radioactive, les différents isotopes du césium conduisent à des isotopes stables du baryum :
Pendant les 300 premières années après déchargement du combustible des réacteurs, la radiotoxicité du césium domine celle des produits de fission présents dans le combustible. Après 300 ans de déchargement, le 137Cs a quasiment disparu et seul subsiste pendant des millions d'années l'isotope radioactif 135Cs qui émet uniquement des particules β d'énergie 0,205 MeV. Avec l'ensemble des autres radionucléides présents dans les solutions de produits de fission (alcalino-terreux, métaux de transition, lanthanides, actinides…), le césium est actuellement immobilisé au sein du réseau vitreux de matrices borosilicatées (verre R7T7 en France, qui renferme 1,29% de Cs2O en masse). Ce type verre a été choisi pour sa capacité à accueillir toute la diversité des produits de fission, des actinides (Np, Am, Cm) et des autres éléments présents dans ces solutions. Il n'a donc pas été optimisé pour le confinement spécifique du césium.
1.2 Intérêt de la recherche de matrices de confinement spécifiques pour le césium
En raison de la forte mobilité du césium dans l'environnement, de sa solubilité élevée dans l'eau et donc des risques potentiels de passage de 135Cs (radionucléide à vie très longue) dans la biosphère à long terme, d'autres types de matrices permettant d'accueillir des quantités plus importantes de césium et plus durables chimiquement que les verres borosilicatés actuels ont été envisagées pour son confinement spécifique. Pour cela, il a été montré qu'une séparation préalable du césium à partir des solutions de déchets nucléaires hautement radioactives à l'aide de molécules extractantes (calixarène-couronnes) très sélectives vis à vis des autres produits de fission était possible [5,6]. En outre, il est important de noter que 137Cs est responsable d'un important dégagement de chaleur lors des premières années suivant le déchargement du réacteur. En effet, pour une tonne d'uranium irradié à un taux de combustion de 33000 MWj/t à la sortie du réacteur, sa contribution dans les solutions de produits de fission, atteint 46% de la puissance dégagée par l'ensemble des produits de fission et des actinides après 50 ans de refroidissement [7]. De ce fort pouvoir thermogène résulte une limitation du taux d'incorporation du césium dans les matrices de confinement spécifiques à 5% massiques d'oxyde de césium Cs2O. Au-delà de cette gamme de concentration, la température au cœur de la matrice dépasserait 500 °C ce qui entraînerait des difficultés d'entreposage et de stockage des colis de déchets (nécessité de refroidissement important).
Une matrice envisageable pour immobiliser spécifiquement le césium doit donc satisfaire à différents critères résultant des propriétés propres au césium mais aussi de la faisabilité technique de la synthèse du matériau. Ainsi, les principaux points à considérer lors du choix d'une matrice de confinement pour le césium sont les suivants :
- - Cette matrice doit présenter une très bonne capacité d'accueil du césium et de son fils le baryum produit au cours de sa décroissance radioactive. Notons également que cette matrice devra accueillir non seulement le césium (rCs+ (8) = 1,74 Å) et le baryum (rBa2+ (8) = 1,42 Å) mais aussi le rubidium (rRb+ (8) = 1,61 Å), co-extrait lors de la séparation du césium par les calixarène-couronnes, et quelques traces de strontium (rSr2+(8) = 1,26 Å) issues de la désintégration du rubidium radioactif (les valeurs des rayons ioniques données ici pour les différents cations [2] correspondent à la coordinence 8 qui est celle des sites d'accueil de ces ions dans les matrices de type hollandite).
- - En raison des risques d'élévation de température au cours du stockage, cette matrice doit aussi posséder une forte stabilité thermique.
- - Cette matrice doit présenter une excellente durabilité chimique à long terme, c'est-à-dire sur des millions d'années. Cette propriété - qui reflète la très faible tendance de la matrice à se solubiliser dans l'eau - limite les risques de relâchement du césium vers la biosphère.
- - Cette matrice doit posséder une bonne stabilité sous irradiation β et γ provenant de la désintégration du césium radioactif. Ces irradiations engendrent généralement l'échauffement du matériau et la création de défauts électroniques ainsi que structuraux via l'excitation, l'ionisation du matériau et des chocs balistiques. La matrice doit conserver au cours du temps ses performances, notamment en terme de rétention du césium et de durabilité chimique, malgré les dégâts induits.
- - Cette matrice doit pouvoir être synthétisée en un nombre limité d'étapes et à un coût raisonnable. Le césium étant labile et volatil impose de plus d'autres critères d'élaboration. La volatilité du césium limite le traitement thermique en température et en durée de la synthèse de la matrice. De plus, comme le césium a tendance à former des phases parasites hydrosolubles, il est nécessaire d'obtenir une matrice monophasée ayant incorporé le plus possible du césium introduit avant synthèse.
1.3 Les matrices de structure hollandite pour le confinement spécifique du césium radioactif
Parmi les différentes matrices vitreuses ou céramiques qui ont été envisagées pour immobiliser le césium, les matrices céramiques de structure hollandite (BaxCsy(M,Ti)8O16, x + y < 2 et M cation trivalent) apparaissent aujourd'hui comme les meilleures candidates et en particulier des compositions pour lesquelles M = Fe3+ ou Fe3+ + Al3+ (BaxCsy(Fe,Al)3+2x+yTi4+8−2x−yO16) qui conduisent aisément (sans à avoir à utiliser des conditions réductrices lors de la synthèse) à l'accueil de quantités significatives d'ions Cs+ au sein de leur structure [8–12]. Ces matrices hollandite sont en effet envisagées comme matrices spécifiques pour le césium à la fois par l'ANSTO (Australian Nuclear Science and Technology Organisation) en Australie [13] et par le CEA en France [8]. Ce type de matrice peut incorporer plus de 5% massiques d'oxyde de césium, accepte dans sa structure le produit de décroissance du césium radioactif (baryum), la présence d'ions Ti4+ dans la structure permet de piéger les électrons émis lors de la désintégration β du césium (Ti4+ + β → Ti3+) et possède une durabilité chimique supérieure d'environ 2 ordres de grandeur à celle des verres borosilicatés [8,14–16]. La formule générale de la hollandite est A2B8O16 dans laquelle les cations A peuvent être le césium, le rubidium et le baryum et les cations B regroupent notamment le titane, l'aluminium et le fer. Ce type de matrices, en particulier celles pour lesquelles A = Ba, Cs et B = Al, Ti, a été largement étudié par l'ANSTO dans les années 70–80 dans le cadre de ses recherches sur les matrices céramiques appelées Synroc [17].
La structure de la hollandite (Fig. 1) est constituée de chaînes infinies d'octaèdres BO6 liés par leurs arêtes (O(1)–O(2)) dans la direction de l'axe c pour une hollandite quadratique (ce qui est le cas pour les compositions de hollandite étudiées ici qui contiennent de gros cations dans le site A, en revanche dans le cas de compositions de hollandite renfermant des cations de faible taille, la structure est monoclinique) [18]. Ces chaînes se groupent par deux avec mise en commun de leurs arêtes de type O(1)–O(1). Ces doubles chaînes s'associent ensuite entre elles en partageant un sommet des octaèdres de type O(2). Ce réseau tridimensionnel aménage alors de larges tunnels parallèles à l'axe c, dans lesquels sont situés les cations A (Ba2+ et Cs+) au centre d'un prisme à base carré d'atomes d'oxygène (8 atomes O(1)) (Fig. 1b). La nature des cations B contrôle la taille des tunnels et conditionne l'incorporation des cations A au sein de la hollandite. Ainsi, l'introduction de gros cations (Cs, Rb) dans les tunnels est facilitée si des cations B de rayon ionique élevé sont présents dans les sites octaédriques. Par ailleurs, les polyèdres de coordination de tous les cations de la hollandite sont relativement déformés, en raison de la position décentrée des cations A et B. En pratique, le site A n'est que rarement totalement occupé dans la structure hollandite. Certes, dans le cas des cations A monovalents, le taux de remplissage x (formule de la hollandite AxB8O16) peut atteindre 2 mais pour des cations divalents (tels que le baryum) les tunnels ne sont que partiellement occupés. Le taux d'occupation x maximal dépend de la nature des cations A et B. En effet, des lacunes sont nécessaires pour l'incorporation dans la structure de gros cations A comme le césium. Ces cations se situent de préférence dans des sites voisins de lacunes afin de limiter les distorsions locales. Notons que les cations B trivalents (Al3+, Ti3+, Fe3+) présents dans les sites octaédriques permettent de compenser la charge positive des cations A (Ba2+, Cs+) présents dans les tunnels.
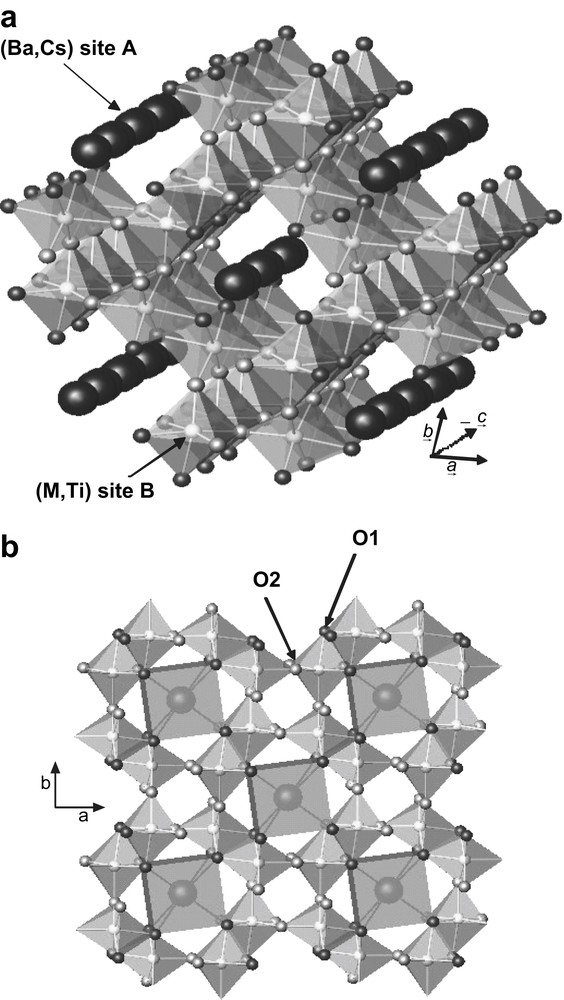
Vue en perspective (a) et projection (b) dans le plan perpendiculaire à l'axe c de la structure quadratique de la hollandite A2B8O16. Les deux types d'oxygène O1 et O2 sont indiqués. M : cations trivalents. En réalité pour les matrices de structure hollandite envisagées dans cette étude, les sites A des tunnels ne sont pas complètement occupés (présence de lacunes coexistant avec les ions Ba2+ et Cs+).
Bien que les matrices hollandite à l'aluminium et au fer évoquées plus haut remplissent l'essentiel des points du cahier des charges d'une bonne matrice pour confiner le césium, les études reportées dans la littérature concernant la tenue de ce type de matrice à l'auto-irradiation due au césium radioactif (irradiation β et γ) demeurent très limitées et jusqu'à ces dernières années aucune étude structurale fine n'avait été menée sur des échantillons de hollandite irradiée. De plus, à notre connaissance aucune expérience de fabrication de hollandite avec du césium radioactif n'a été réalisée à ce jour, pour évaluer sa tenue réelle à l'auto-irradiation à la différence de ce qui a pu être fait pour la pollucite CsAlSi2O6 et certains verres par exemple [19,20]. En outre, aucun analogue minéral naturel ayant contenu du césium radioactif n'existe pour le type de composition de hollandite envisagée ici à la différence de ce qui peut être le cas pour certaines matrices envisagées pour confiner spécifiquement les actinides par exemple (zirconolite, zircon, apatite, monazite). Seules des irradiations externes avec des particules α, des neutrons, des ions lourds ou des électrons issus d'un microscope électronique en transmission ont été reportées sur des échantillons de hollandite pure ou sur des échantillons de Synroc [21–23]. Les essais d'irradiation externe aux électrons issus d'un microscope surestiment les effets d'irradiation engendrés par les particules β émises par le césium radioactif, en raison du fort débit de dose appliqué et de l'absence de recuit potentiel des défauts d'un point de vue cinétique [21,22]. Par conséquent, la stabilité sous irradiation de la hollandite n'avait pas été démontrée à ce jour au travers de ces différentes études.
L'objectif du présent papier est de présenter les principaux résultats que nous avons obtenus par résonance paramagnétique électronique (RPE) et résonance magnétique nucléaire (RMN) sur l'effet d'irradiations électroniques externes (produites par un accélérateur Van de Graaff à une température proche de l'ambiante) simulant dans une certaine mesure les rayonnements β et γ émis par les isotopes radioactifs du césium sur la structure locale d'une hollandite de composition simplifiée Ba1,16Al2,32Ti5,68O16 sans césium. Cette céramique a été préparée par voie oxyde (mélange de poudres) et frittée sous air. De plus, étant donné l'échauffement prévu de la hollandite pendant les premières années de stockage du césium radioactif en raison de la présence de 137Cs (T ∼ 300 °C au cœur du colis des déchets), l'étude de l'évolution thermique (stabilité) des défauts ponctuels primaires formés et détectés par RPE (centres à électrons et centres à trous électroniques) est particulièrement importante et a été réalisée après traitement thermique des échantillons irradiés à température proche de l'ambiante. Ces différents résultats seront discutés au regard de la structure électronique calculée pour la hollandite (nature des centres à électrons et à trous électroniques crées par interaction inélastique) et aux sections efficaces de déplacement calculées pour les différents atomes (interaction élastique électrons-atomes).
2 Protocole expérimental
2.1 Choix de la composition de hollandite étudiée et méthode de synthèse
Bien que les compositions de hollandite actuellement retenues pour le confinement du césium soient des hollandites ferrifères du type BaxCsy(Fe,Al)3+2x+yTi4+8−2x−yO16, pour la présente étude il a été nécessaire d'éviter la présence de fer (paramagnétique) aussi bien pour la RMN que pour la RPE. En effet, la présence d'une forte concentration d'ions Fe3+ conduit à un déplacement et à un élargissement des signaux RMN. De plus, cherchant à détecter des signaux RPE associés à des espèces paramagnétiques induites par irradiation électronique ou γ, la présence d'un signal intense préexistant associé aux ions Fe3+ serait fortement gênante. En raison de la difficulté d'incorporer des quantités significatives de césium dans les tunnels de la hollandite en l'absence de fer et lors d'une synthèse sous air, nous avons donc choisi d'étudier une composition de hollandite sans fer et sans césium (Ba1,16Al2,32Ti5,68O16) dérivée de celle retenue actuellement par le CEA [8]. Notons toutefois que quelques essais d'irradiation électronique menés sur des compositions de hollandite renfermant du césium ou du fer [10] nous ont révélés des défauts paramagnétiques de natures identiques à celles des défauts produits dans la présente étude ce qui permet donc d'étendre les résultats présentés ici au cas des céramiques hollandite ferrifères retenues pour l'immobilisation du césium.
Les méthodes de synthèse des échantillons des céramiques hollandite au césium développées aussi bien en France par le CEA qu'en Australie par l'ANSTO reposent sur le frittage d'un mélange de précurseurs obtenus par voie alcoxyde (alcoxydes (Al, Ti) + nitrates (Ba, Cs, Fe) dans le cas des compositions retenues en France [8,24]). Pour notre étude, les échantillons de céramique hollandite Ba1,16Al2,32Ti5,68O16 ont été synthétisés par voie oxyde (réaction à l'état solide d'un mélange de poudres finement broyées) en partant des matières premières suivantes : BaCO3, TiO2 et Al2O3. Les conditions de synthèse mises en oeuvre ici ont été mises au point pour des échantillons de hollandite au césium dans le but de limiter les envols durant les traitements thermiques et cette méthode de synthèse constitue une alternative à la méthode alcoxyde [25]. Après calcination à 810 °C (4 h) nos échantillons ont été finement broyés (<1 μm) par attrition (en milieu aqueux à l'aide de billes de diamètre compris entre 0,6 et 0,8 mm à base de zircone) puis frittés sous air à 1320 °C (96 h) (Fig. 2). Notons que pour des échantillons renfermant du césium, la température de frittage peut être abaissé à 1200 °C et la durée abaissée à 30 h afin de limiter au maximum le départ de césium au cours de la synthèse. A l'exception de quelques traces d'une phase parasite au niveau des joints de grains contenant du baryum et du phosphore (impureté fréquemment présente dans TiO2) détectées par microscopie électronique à balayage couplée à l'analyse X (Fig. 2), les échantillons apparaissent monophasés en diffraction des rayons X (Fig. 3). La composition déterminée par microsonde électronique (Ba1,17Al2,28Ti5,71O16) est en outre très proche de la composition visée et les échantillons obtenus sont relativement denses (∼90% de la densité théorique). Bien que nous ne connaissions pas l'aptitude de la phase parasite à base de phosphore et de baryum à incorporer du césium, ni son comportement sous irradiation électronique, il est important que de noter que d'une part cette phase est présente en très faible quantité dans nos échantillons et que d'autre part, en nous procurant du TiO2 exempt de phosphore auprès de fournisseurs spécialisés, il nous a été possible de synthétiser ultérieurement des céramiques hollandite sans cette phase parasite [9].
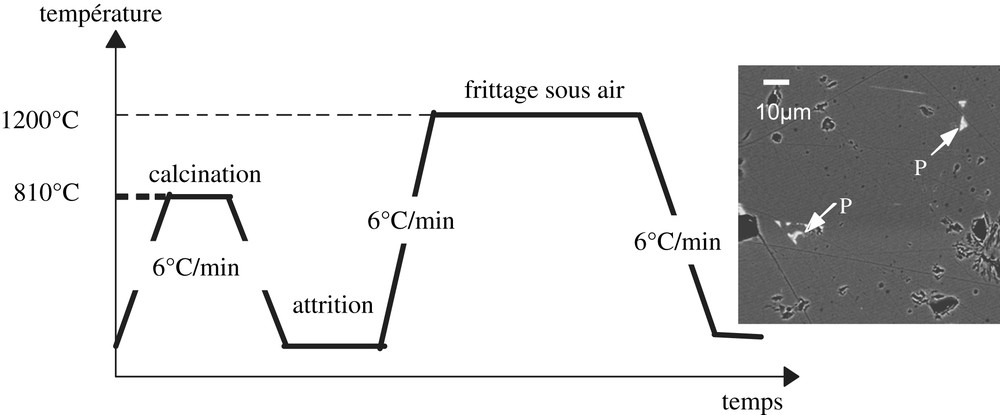
Procédé mis oeuvre pour la synthèse de céramiques hollandite par voie oxyde : calcination sous air (4 h) de pastilles pressées à froid, broyage par attrition et frittage naturel (30 h) de pastilles pressées. Un cliché obtenu par microscopie électronique à balayage (électrons rétrodiffusés) de la céramique Ba1.16Al2.32Ti5.68O16 est également présenté. Les zones sombres correspondent à des porosités et la phase blanche très minoritaire correspond à une phase parasite P (de structure non identifiée) riche en phosphore.
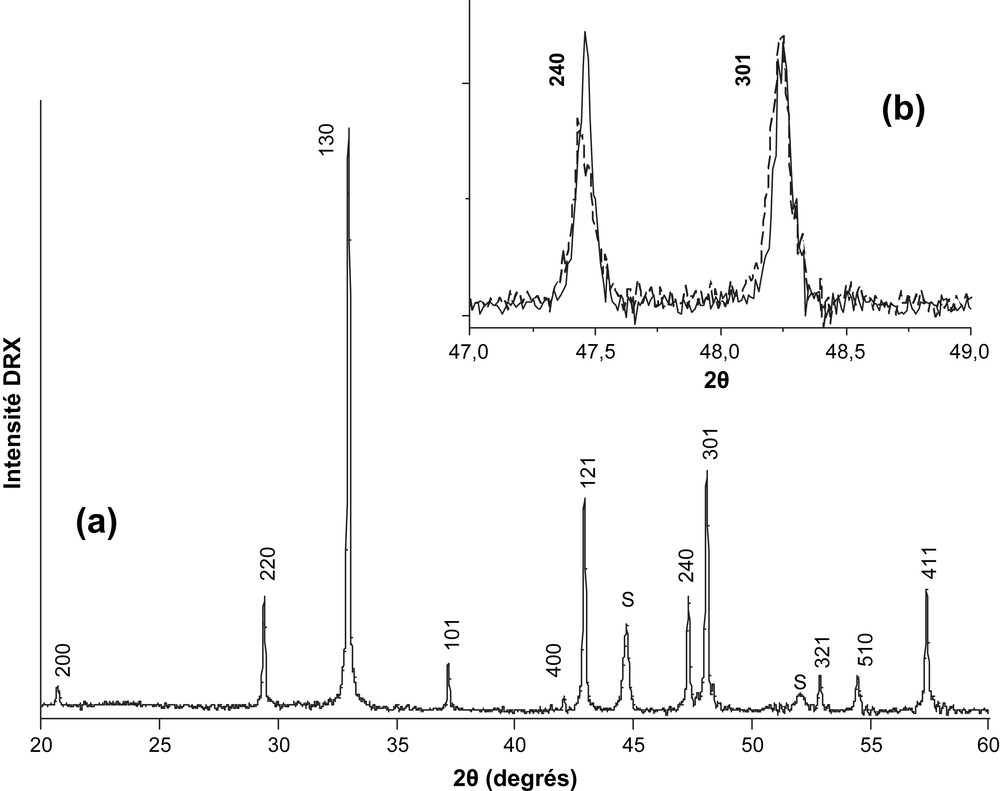
(a) Diagramme de diffraction des rayons X de la hollandite céramique Ba1.16Al2.32Ti5.68O16 préparée par voie oxyde à 1200 °C. Toutes les raies ont été indexées dans le groupe d'espace I4/m (structure tétragonale). S : support an aluminium. (b) Détail du diagramme montrant un léger élargissement des raies après irradiation électronique (énergie : 2,5 MeV, fluence : 1,2 × 1019 cm−2, courbe en pointillés) attribuable à une augmentation du désordre au sein de la structure (λ Co Kα1 = 1,78897 Å).
2.2 Irradiation électronique externe des échantillons de hollandite
Pour simuler l'effet de la radioactivité β du césium radioactif, différents échantillons de hollandite Ba1,16Al2,32Ti5,68O16 ont été irradiés de façon externe par des électrons générés à l'aide d'un accélérateur Van de Graaff produisant des électrons d'énergie E comprise entre 1 et 2,5 MeV (c'est à dire à des énergies supérieures à la majorité des particules β émises par 137Cs et 135Cs, Fig. 1), une fluence F comprise entre 3,4 × 107 et 1,4 × 109 cm−2 et un débit de dose important (2–7 × 106 Gy h−1) permettant d'obtenir en quelques jours une dose absorbée de l'ordre de 109 Gy, correspondant à environ 30 années de stockage des déchets radioactifs. Une étude de l'influence des différents paramètres décrivant les irradiations tels que la fluence (nombre d'électrons/cm2), la dose absorbée (énergie absorbée par unité de masse du matériau) et le nombre de déplacements atomiques a été réalisée. Afin de mener à bien cette étude, différents logiciels d'irradiation (ESTAR, PENELOPE) ont été utilisés [10]. De plus, les sections efficaces σd de déplacement des différents éléments constitutifs de la hollandite ont été calculés à l'aide du logiciel développé par Dunlop et al. [26] traitant des collisions primaires et secondaires engendrées par un électron sur un atome donné. En effet, les collisions élastiques, bien qu'elles ne soient pas prépondérantes dans le cas des irradiations électroniques en raison de la faible masse des électrons, ne doivent pas être négligées dans cette étude car certaines techniques de caractérisation des effets d'irradiation (RPE par exemple) permettent de détecter les défauts ponctuels pouvant provenir notamment de déplacements atomiques. Nous avons déterminé la section efficace de déplacement σd de chaque élément en utilisant des valeurs de Ed (seuil de déplacement), aussi bien pour l'oxygène que pour les cations, comprises entre 20 et 60 eV (Fig. 4). La variation de la section efficace de déplacement des ions en fonction de l'énergie incidente des électrons et de l'énergie de déplacement Ed de l'ion considéré montre que les atomes légers (O, Al) de la matrice commencent à se déplacer à plus faible énergie que les atomes plus lourds (Ti, Ba). Par contre, leurs sections efficaces de déplacement sont moins importantes que celle des atomes lourds et notamment de celle du baryum à forte énergie. En comparant la valeur des sections efficaces aux différentes énergies incidentes utilisées, nous pouvons conclure que tous les atomes de la hollandite peuvent être déplacés lors des irradiations électroniques à 2,5 MeV quelles que soient les valeurs d'énergie de déplacement considérées (entre 20 et 60 eV). Par contre, pour des irradiations à 1 et 1,5 MeV, les atomes de baryum ne seront pas déplacés si leur énergie de déplacement Ed est supérieure à 25 eV ou 50 eV, respectivement. Par conséquent, si un effet de seuil est observé entre les différentes irradiations de 1 et 2,5 MeV, les atomes de baryum en seront responsables.

Evolution de la section efficace de déplacement en fonction de l'énergie des électrons incidents (0–2,5 MeV) et de la valeur de l'énergie de déplacement Ed (20–60 eV) pour le baryum (a) et l'oxygène (b) obtenue à l'aide du programme développé par Dunlop et al. [26].
Lors des irradiations, les échantillons de hollandite ont été disposés sur un socle en cuivre qui permet de les maintenir à une température proche de l'ambiante pendant l'irradiation (T ∼ 50 °C). Les échantillons se présentent sous la forme de fins rectangles obtenus après découpe et polissage des échantillons massifs de céramique. Leurs dimensions sont les suivantes : longueur comprise entre 5 et 10 mm, largeur de 4 mm, épaisseur de 0,5 mm à 1 mm selon l'énergie des électrons incidents. La surface irradiée est comprise entre 20 et 40 mm2. L'épaisseur des échantillons a été choisie inférieure au parcours moyen R des électrons incidents calculé à l'aide du logiciel ESTAR [10] pour que les électrons traversent l'échantillon, évitant ainsi tout problème de charge du matériau isolant. Par conséquent ces irradiations externes ne simuleront que l'effet des rayonnements β le long de leur parcours dans la hollandite. Les épaisseurs des échantillons, relativement faibles par rapport à R ont été sélectionnées pour obtenir des effets d'irradiation relativement homogènes dans tout le volume, c'est à dire une énergie déposée par particule et par mm assez similaire tout le long du parcours de l'électron incident. Ce calcul a été effectué à l'aide du logiciel PENELOPE [10]. La Fig. 5 montre que les photons γ émis par 137Cs interagissent préférentiellement par effet Compton avec la hollandite (le numéro atomique effectif Zeff de la hollandite de composition Ba1,16Al2,32Ti5,68O16 étant estimé à 38 [10]). Des électrons Compton d'énergie pouvant atteindre 0,477 MeV sont alors générés par les rayons γ issus de 137Cs (0,662 MeV). Ces électrons Compton génèreront donc les mêmes dégâts d'irradiation dans la hollandite que les particules β émises par 137Cs (Fig. 1). On peut donc considérer que les irradiations électroniques externes simulent également l'effet des rayons γ sur nos échantillons de hollandite.
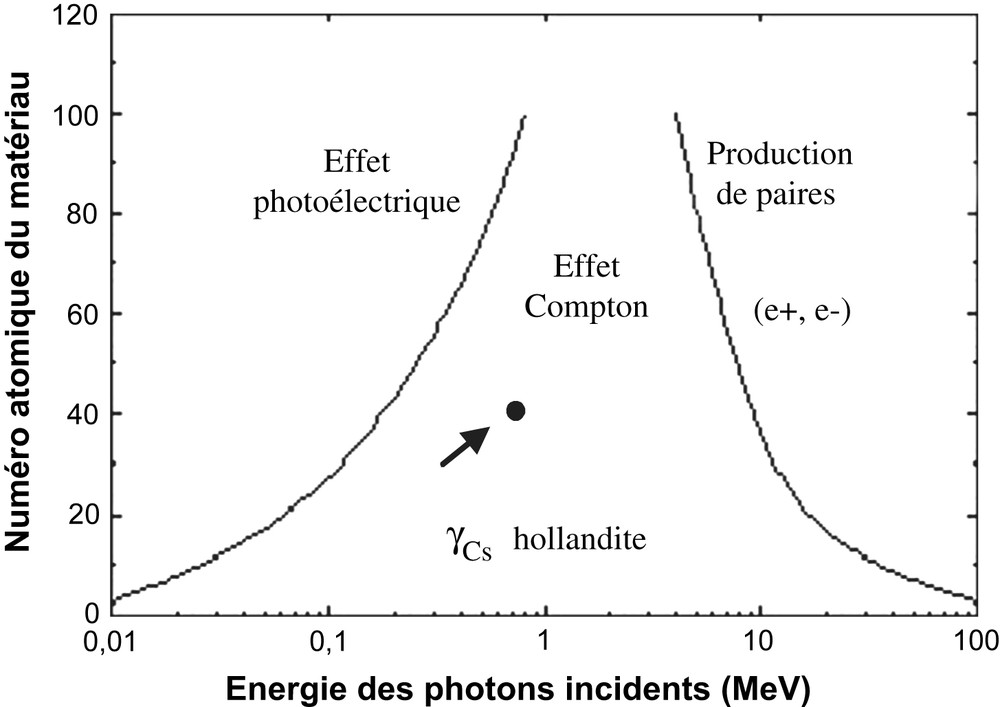
Importance relative des 3 processus d'interactions possibles des photons γ en fonction de leur énergie et du numéro atomique effectif du matériau cible (d'après [27]). Le point correspondant à la hollandite étudiée ici et à l'énergie des photons γ émis par 137Cs est indiquée et montre que ceux-ci interagissent préférentiellement par effet Compton avec la hollandite.
Comme les irradiations électroniques induisent principalement des ionisations et des excitations électroniques (création de paires électrons-trous électroniques) dans les matériaux, la formation de défauts ponctuels paramagnétiques est attendue dans la hollandite. Afin de prévoir la nature de ces défauts nous avons réalisé un calcul semi-empirique de la structure de bande de la hollandite stoechiométrique BaAl2Ti6O16 à l'aide d'une méthode de Hückel étendue [10] (Fig. 6a).
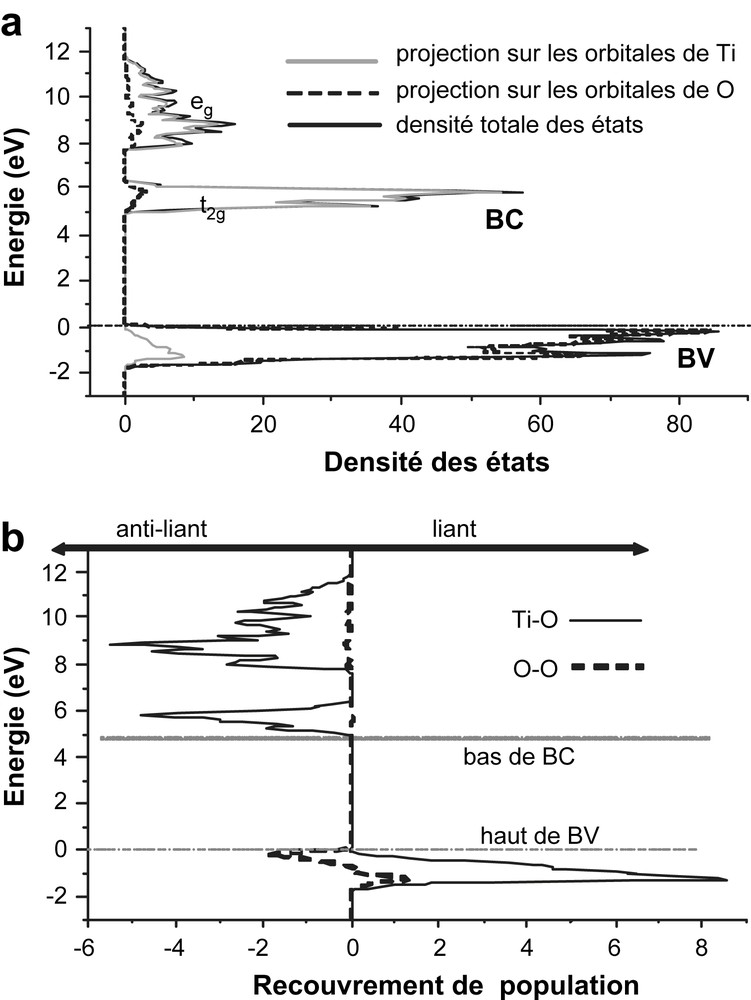
(a) Structure de bande autour du gap de la hollandite de composition BaAl2Ti6O16 représentant l'énergie des états en fonction de leur densité totale ou de leur densité projetée sur les atomes d'oxygène ou sur les atomes de titane. (b) Densité de populations de recouvrement entre les différents atomes de la hollandite de composition BaAl2Ti6O16 en fonction de l'énergie des états.
Il apparaît que le haut de la bande de valence et le bas de la bande de conduction sont principalement constitués d'orbitales 2p des atomes d'oxygène et des orbitales 3d des atomes de titane respectivement. Par conséquent, on s'attend à ce que les centres à électrons soient des ions Ti3+ ions (après piégeage des électrons par les ions Ti4+) et les centres à trous des centres de type O− ou O2n− (n < 4) (après piégeage d'un ou de plusieurs trous par les ions oxygène). La liaison O–O étant anti-liante à l'extrême haut de la bande de valence comme l'illustre la Fig. 6b représentant les densités de population de recouvrement selon l'énergie des états de la hollandite. Le piégeage d'un trou électronique engendre alors une diminution de la distance O–O (renforcement du caractère liant de la liaison) et ainsi la formation d'espèces du type O23−. Si plusieurs trous sont piégés, des espèces O2− peuvent alors être créées.
L'étude RPE a été réalisée sur les échantillons de hollandite (irradiés et irradiés + recuits) à l'aide de spectromètres Bruker ESP 300e et ELEXYS E500 (9,5 GHz) entre 9 et 300 K. Pour plus de détails concernant la méthodologie mise en oeuvre pour l'exploitation des spectres RPE voir [28]. Afin d'étudier d'une part la stabilité des défauts paramagnétiques en fonction de T et de tenir compte du fait qu'au début du stockage la température au coeur des colis sera de l'ordre de 300 °C (immobilisation de 5% Cs2O en masse), des traitements thermiques isochrones (15 min) et des traitements isothermes (jusqu'à 215 h) ont été réalisés sur un échantillon irradié. Les spectres RMN 27Al (rotation à l'angle magique simple (MAS) et multiquanta (3Q-MAS)) des échantillons avant et après irradiation ont été enregistrées à l'aide d'un spectromètre BRUKER AVANCE 500 (11.7 T) [28]. Les déplacements chimiques sont reportés par rapport à AlCl3·6H2O.
3 Résultats et discussion
3.1 Modifications structurales induites dans la hollandite par irradiation électronique
L'étude par diffraction X des échantillons de hollandite Ba1,16Al2,32Ti5,68O16 irradiée (E = 2,5 MeV, F = 1,2 × 1019 cm−2) ne révèle qu'un très faible élargissement des pics de diffraction (Fig. 3b). traduisant ainsi une légère augmentation du désordre après irradiation. En outre, la comparaison des diagrammes de diffraction électroniques et des clichés de microscopie électronique en transmission à haute résolution avant et après irradiation (E = 1,5 MeV, F = 5,8 × 1018 cm−2) mettent en évidence un déplacement des ions Ba2+ dans les tunnels sous l'effet de l'irradiation électronique. Trois environnements locaux des ions Al3+ en site octaédriques (sites S1, S2 et S3) ont été mis en évidence par RMN dans la hollandite avant irradiation, différant par l'arrangement des cations baryum les plus proches dans les tunnels [10]. Après irradiation (Fig. 7a), aucune évolution significative concernant les proportions de ces différents sites n'a été observée par conséquent aucune modification de l'arrangement des cations Ba2+ autour des ions Al3+ n'est observée par RMN. Cela montre que la RMN ne possède pas une assez grande sensibilité pour détecter ces effets, des déplacements de baryum étant mis en évidence par diffraction électronique et par microscopie électronique en transmission sur ces échantillons. De plus, l'absence d'élargissement du signal suggère que la concentration en espèces paramagnétiques créées par les irradiations électroniques (telles que des ions Ti3+, comme cela est attendu) demeure très faible, tout au moins inférieure à 1 ‰ des cations B. Par contre, un faible épaulement au signal RMN, indiqué par une flèche dans la Fig. 7b, est induit par les irradiations électroniques. Ce nouveau signal est situé autour de +45 ppm, correspondant à la gamme des déplacements chimiques de l'aluminium en coordinence V et IV. Ce signal possède des temps de relaxation similaires aux signaux S1, S2 et S3, ce qui suggère qu'il est dû à des ions Al3+ dans la structure hollandite et non dans une phase parasite. Il représente environ 5% de la totalité des ions Al3+ pour l'échantillon irradié à E = 1 MeV. Ainsi, le faible effet mis en évidence dans la Fig. 7b indique que les irradiations électroniques engendrent la formation d'ions Al3+ en coordinence réduite résultant de la formation de lacunes d'oxygène.
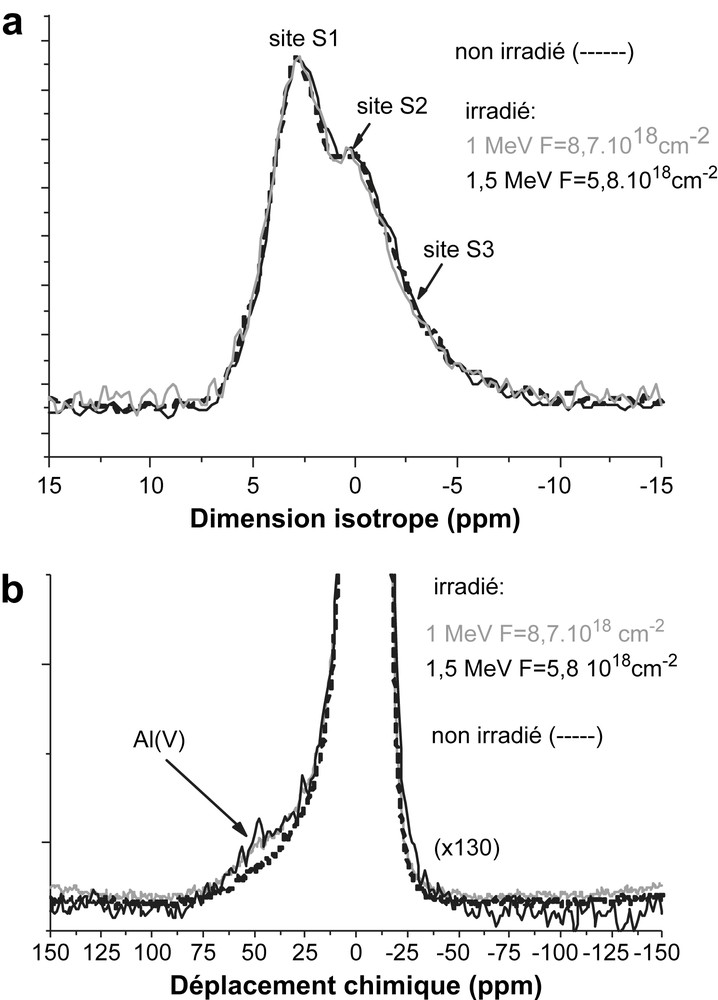
(a) Projection dans la dimension isotrope du spectre 3Q-MAS avant et après irradiation de la hollandite Ba1,16Al2,32Ti5,68O16 (b) Spectres RMN MAS 27Al avant et après irradiation. L'intensité des spectres sur une même figure a été ramenée à la même échelle.
Pour confirmer ces modifications détectées par RMN de 27Al, nous avons également caractérisé les effets des irradiations électroniques par RMN MAS de 17O sur une hollandite de même composition mais enrichie en 17O [10]. Les spectres RMN MAS de 17O avant et après irradiation tendent aussi à montrer que les irradiations électroniques affectent les ions oxygène. Par conséquent, cette étude RMN montre que les irradiations électroniques induisent la formation de lacunes d'oxygène autour des ions Al3+ de la hollandite. Cela est par ailleurs en accord avec les résultats que nous avons obtenus par spectroscopie Mössbauer du fer pour une hollandite Ba1,16Fe2,32Ti5,68O16 irradiée [10,29].
3.2 Etude RPE de la hollandite irradiée
Comme le montre les spectres de la Fig. 8, les irradiations électroniques réalisées sur la hollandite Ba1,16Al2,32Ti5,68O16 engendrent la formation de 3 signaux Tr, E1 et E2.

Influence de l'énergie E des électrons (a) et de la fluence F des électrons à 1 MeV (b) sur les signaux RPE induits (enregistrement à T = 70 K).
Le signal Tr situé entre 330 et 340 mT et ainsi associé à des facteurs g supérieurs à ge (facteur g de l'électron libre : 2,0023), est attribuable à des centres à trou électronique (espèces oxygénées). Les signaux E1 et E2 localisés à champ plus fort (340–351 mT et 342–370 mT, respectivement) correspondant à des facteurs g inférieurs à ge, sont associés à des centres à électrons (centres Ti3+). Par conséquent, les irradiations électroniques génèrent principalement des centres à trou électronique à base d'oxygène et des centres à électrons de type Ti3+, comme cela était prévisible à partir de la structure de bande (Fig. 6). Ces signaux sont stables dans le temps à température ambiante, jusqu'à deux ans et demi (au moins) après irradiation. Quelles que soient les conditions d'irradiation, seuls ces trois signaux Tr, E1 et E2 sont observés. Néanmoins, leur intensité relative diffère selon l'énergie et la fluence. En effet, comme l'illustre la Fig. 8a, la proportion relative du signal E2 par rapport au signal E1 augmente avec l'énergie tandis que l'intensité du centre Tr par rapport aux signaux des centres à électrons diminue. Une telle influence de l'énergie incidente sur l'intensité du signal E2 suggère qu'il résulte de déplacements atomiques plutôt que de simples excitations électroniques. Par ailleurs, pour une énergie donnée (1 MeV ou 2,5 MeV), l'évolution des intensités relatives des signaux E2 et Tr en fonction de la fluence est également caractérisée par un augmentation de l'intensité relative du signal E2 et une diminution de la proportion relative du signal Tr lorsque la fluence augmente (Fig. 7b). Le signal E2 est attribuable à des déplacements de baryum. En effet, l'évolution de la concentration de spins du signal E2 en fonction de la fluence a été déterminée à diverses énergies [10]. Elle se révèle linéaire avec des pentes, appelées sections efficaces de production σp, différentes selon la valeur de l'énergie des électrons incidents. Un tel effet de l'énergie des électrons confirme que ce signal résulte de déplacements atomiques, qui sont en faibles quantités à 1 MeV, mais deviennent significatifs dès 1,5 MeV. Pour déterminer la nature des atomes déplacés, les rapports des pentes σp(2,5 MeV)/σp(1 MeV) (≈6,2) et σp(1,5 MeV)/σp(1 MeV) (≈3,5) ont été comparés aux rapports des sections efficaces de déplacement des atomes constitutifs de la hollandite, fonction de leur énergie de déplacement Ed (Fig. 4). De telles valeurs ne peuvent pas être atteintes pour les atomes légers tels que l'aluminium ou l'oxygène, au contraire des atomes de titane et de baryum. Ces deux rapports de pentes σp s'accordent parfaitement aux valeurs des rapports des sections efficaces du baryum pour une énergie de déplacement de 21 eV. Cette étude ne nous permet pas de savoir si le signal E1 résulte de déplacements d'éléments légers ou de dépôt d'énergie électronique. Néanmoins, il est certain qu'il ne peut pas être associé à des déplacements de baryum ou de titane (voire d'aluminium). Par ailleurs, cette étude révèle que la concentration des centres à trou électronique est toujours très nettement inférieure à celle des centres à électron (facteur 2 à 13). Cela suggère que d'autres centres à trou électronique diamagnétiques non détectés par RPE existent et/ou que le centre Tr résulte de plusieurs piégeages de trous électroniques successifs.
Ces différents signaux ont été simulés, leurs tenseurs g extraits et identifiés en étudiant en particulier des monocristaux de hollandite irradiée [25,28,30] et comparés avec les données RPE de la littérature. Il apparaît ainsi que les centres à trou électronique correspondent à des ions superoxydes O2− (signal Tr). Nous pensons que ces centres O2− se sont formés par piégeages successifs de trois trous électroniques sur deux ions O2− [28]. Cela pourrait donc expliquer la plus faible concentration en centres à trous qu'en centres à électrons observée plus haut. Les deux centres à électrons sont attribuables à des ions Ti3+ formés dans le volume des grains de hollandite de la céramique : le premier (signal E1) est un ion Ti3+ en coordinence 5, présentant une lacune d'oxygène dans sa sphère de coordination tandis que le second (signal E2) est un ion Ti3+ en coordinence 6 dont l'environnement est enrichi en baryum (Fig. 9). Un mécanisme de formation de ces différents centres a été proposé dans [28] tenant compte à la fois du fait que dans la hollandite, les irradiations électroniques induisent à la fois des excitations électroniques (formation de centres à électrons et à trous) et des déplacements atomiques (ions baryum et oxygène).
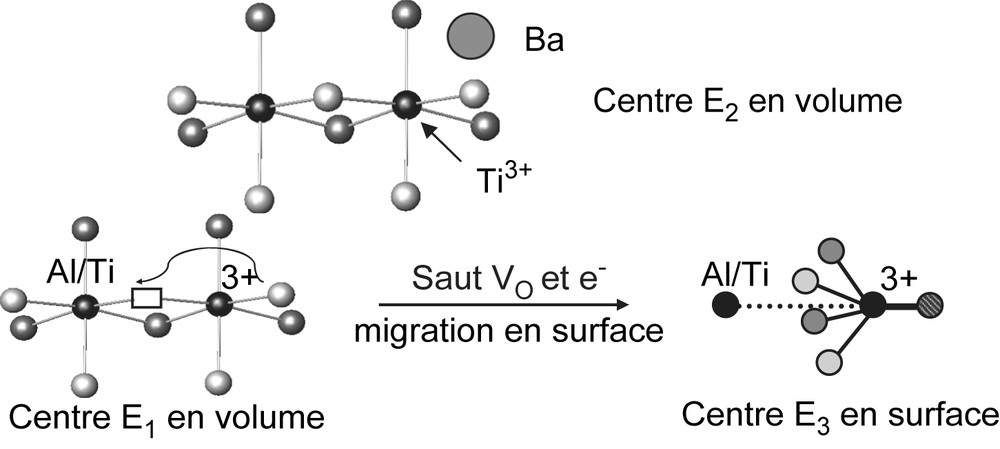
Structure des centres E1 et E2 formés dans le volume des grains de hollandite Ba1,16Al2,32Ti5,68O16 lors de l'irradiation électronique. Le mécanisme de formation des centres E3 (en surface) lors des recuits, par migration simultanée vers la surface des lacunes d'oxygène (VO) et des électrons est également présenté.
Dans la Fig. 10 est présentée l'évolution du spectre RPE d'un échantillon de hollandite irradiée puis soumis à des traitements thermiques isochrones entre 50 et 750 °C. Les signaux E2 et E1 disparaissent à 150 et 300 °C respectivement, et le signal Tr à 350 °C. Ainsi, les centres à trou électronique (Tr) sont plus stables que les centres à électrons, et en particulier que les centres E2. D'autre part, la disparition non simultanée des centres à trou et des centres à électron montre qu'il n'y a pas de mécanisme direct de recombinaison électron-trou entre ces défauts, et suggère donc des mécanismes plus complexes pouvant faire intervenir d'autres défauts diamagnétiques. Par ailleurs, ces traitements thermiques engendrent non seulement la disparition des signaux induits mais aussi l'apparition de nouveaux signaux G2 et E3. Ces signaux n'apparaissent que sur les échantillons irradiés. La Fig. 10 montre qu'il est nécessaire de chauffer la hollandite irradiée jusqu'à 800 °C pour obtenir un spectre RPE relativement similaire à celui d'un échantillon non irradié. Il apparaît donc qu'à 300 °C (température maximale estimée au coeur des colis de hollandite au début du stockage) des défauts provenant de l'évolution des défauts primaires existent toujours et vont probablement s'accumuler au cours du temps. Cependant, il faut noter que les concentrations des différents centres mises en jeu même après les doses d'irradiation les plus fortes restent relativement faibles (<2,3 × 1017 spins/cm3 à comparer aux 2 × 1020 ions titane/cm3 dans la structure de la hollandite). Lors des traitements isothermes à 300 °C, il apparaît que les centres E1 et E2 disparaissent très rapidement (dès 15 min) et les signaux E3 et G2 apparaissent par la suite.
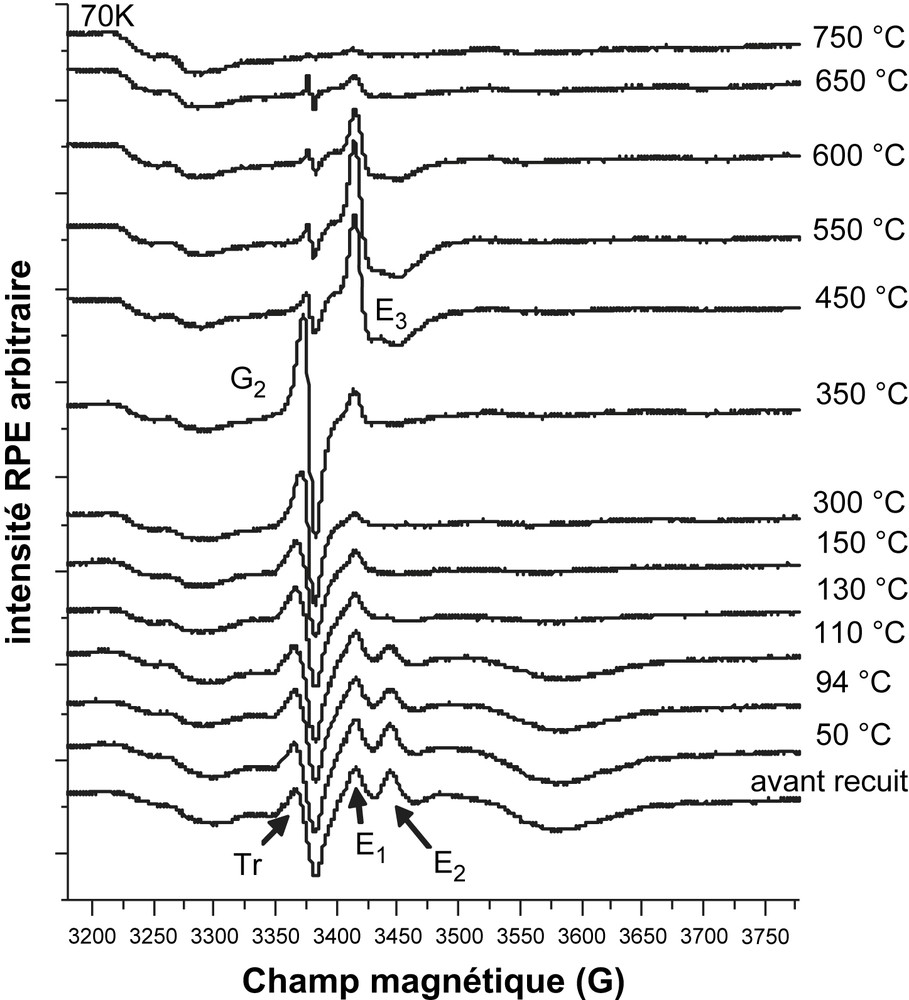
Evolution des spectres RPE d'un échantillon de hollandite irradié E = 1 MeV et F = 1,4 × 1019 cm−2 au cours de traitements thermiques isochrones de 15 min. La température du recuit est indiquée près de chaque spectre (spectres enregistrés à T = 70 K).
Une étude fine des spectres RPE des céramiques irradiées et recuites et de monocristaux traités sous argon [10] nous a permis de montrer que le signal E3 était associé à des ions titanyl de surface avec la liaison courte TiO dirigée selon l'axe c de la structure hollandite, possédant dans leur environnement immédiat soit un ion titane (espèce majoritaire), soit un ion aluminium (espèce minoritaire) (Fig. 9). Le fait que la disparition du signal E1 sous l'effet des traitements thermiques isochrones soit suivie de l'apparition du signal E3 (Fig. 10) suggère un lien de filiation entre les défauts E1 et E3. Comme le signal E1 correspond à des ions Ti3+ de volume possédant une lacune d'oxygène en premier voisin (Fig. 9), nous proposons que le centre E1 est précurseur du centre E3, la transformation s'effectuant par réarrangement des atomes d'oxygène autour du titane, avec migration vers la surface (Fig. 9). Une étude fine du signal G2 nous a permis en outre de proposer que ce signal était attribuable à des agrégats d'espèces oxygénées O22− (diamagnétiques) et O2− (paramagnétiques) de taille inférieure à 10 μm. De tels agrégats pourraient se former lors du traitement thermique des échantillons irradiés par un mécanisme de saut électronique et atomique (Fig. 11). En effet, le transfert thermiquement activé d'un électron d'un ion O2− vers un ion superoxyde voisin aura pour effet d'affaiblir la liaison chimique entre les deux atomes d'oxygène de cette espèce (peuplement d'une orbitale anti-liante, Fig. 6b), favorisant ainsi le saut d'un atome d'oxygène entre deux sites voisins. Une telle migration d'atomes d'oxygène pourrait alors produire une accumulation de ces espèces riches en oxygène mais faiblement chargées à certains endroits du matériau. Une ultime capture de trous électroniques pourrait aboutir à la formation d'oxygène moléculaire O2. Nous n'avons cependant pas observé cette ultime étape par spectroscopie Raman (absence du pic caractéristique de l'oxygène moléculaire, attendu à 1550 cm−1).
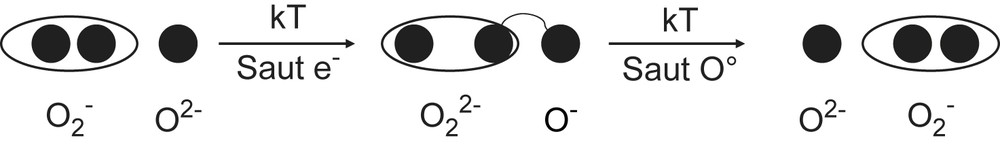
Mécanisme proposé pour la diffusion des centres à trous électroniques (ions superoxyde O2−) lors des recuits conduisant à des agrégats (centres G2).
La Fig. 12 résume l'effet des irradiations électroniques sur un grain de hollandite Ba1,16Al2,32Ti5,68O16 (formation des centres E1, E2 et Tr) et du traitement thermique conduisant aux centres E3 (surface) et G2 (agrégats dans la masse).

Effet des irradiations électroniques suivi de recuits sur un grain de hollandite de la céramique Ba1,16Al2,32Ti5,68O16.
4 Conclusions
Cette étude a donc montré que la hollandite est un matériau globalement résistant aux irradiations électroniques, tant au niveau de sa structure que de la faible concentration en défauts ponctuels induits. Les irradiations ont uniquement induit des modifications de l'environnement local des cations du squelette (Al3+ et Ti4+), résultant d'une augmentation du désordre dans les tunnels, associée à des déplacements d'ions baryum et à la formation de lacunes d'oxygène. La RPE a notamment mis en évidence les défauts ponctuels relatifs à ces modifications autour du titane tels que des ions Ti3+ dont l'environnement est enrichi en baryum (centre E2) et des ions Ti3+ associés à une lacune d'oxygène (centre E1). Des espèces oxygénées de type O2− (centre Tr) sont également formées. Ces défauts sont relativement stables dans le temps (au moins deux ans et demi à température ambiante) et en température (disparition entre 150 et 350 °C lors de traitements thermiques isochrones de 15 min). De plus, leur recuit thermique engendre la formation d'autres défauts correspondant à des ions Ti3+ de surface de type titanyl (centre E3) et des agrégats d'oxygène (centre G2). L'apparition de ces centres indique une séparation des centres à électron et des centres à trou électronique, ce qui suggère qu'à terme ces défauts vont s'accumuler au lieu de se recombiner, pouvant alors engendrer des modifications plus importantes du matériau (Fig. 12).
Toutefois il est important de souligner les différences entre les irradiations électroniques externes réalisées dans le cadre de notre étude et les conditions réelles d'auto-irradiation lors du stockage du césium radioactif. Il apparaît ainsi que ces irradiations externes simulent principalement les défauts produits par les particules β le long de leur parcours dans les grains de hollandite. De plus, dans la présente étude, l'énergie des particules β, le débit de dose et la température d'irradiation sont différents des conditions réelles. Tout d'abord, la désintégration β du césium produit essentiellement des électrons de 0,514 MeV (137Cs) qui n'engendreront pas de déplacements de baryum, d'après l'énergie de déplacement des ions baryum estimée à 21 eV (Fig. 4). Par conséquent, le défaut E2 ne sera pas formé en grande quantité lors du stockage. De plus, certaines des modifications de l'environnement local des ions Al3+, résultant d'un réarrangement des cations Ba2+, ne seront probablement pas induites. Néanmoins, la désintégration β engendrera des déplacements d'atomes d'oxygène et des excitations électroniques. Par conséquent, les défauts de type Ti3+ (E1) et O2− seront probablement formés. Or, ce sont ces derniers qui conduisent à la formation d'agrégats d'oxygène qui pourraient induire des modifications à long terme du matériau. De plus, ces agrégats sont formés lors des recuits isothermes à 300 °C des matériaux irradiés et présentent une grande stabilité dans le temps. Par ailleurs, nous avons vu que nos irradiations ne simulent que les 30 premières années de stockage et donc une éventuelle accumulation de ce type de défauts est à attendre. Par conséquent, un des seuls points qui pourrait jouer un rôle sur la résistance aux irradiations de la hollandite est la présence de ces agrégats. Il serait alors important d'effectuer des irradiations directement à haute température (300 °C) pour déterminer si ce défaut de type agrégats est également formé dans ces conditions.
Cependant, l'ensemble de nos résultats tend à montrer que la structure hollandite est résistante à l'irradiation électronique simulant ici le rayonnement β émis par les ions césium radioactifs car les défauts qui persisteront à long terme ne représenteront qu'une très faible fraction du matériau. Signalons également que des travaux récents [31] d'irradiation électronique externe réalisés au sein d'un microscope électronique à transmission sur des échantillons de hollandite de composition Ba0,85Cs0,26Al1,35Fe0,77Ti5,90O16 tendent à confirmer la stabilité de la structure hollandite sous rayonnement β. De plus, dans ces mêmes travaux, aucun effet d'une irradiation γ (2,4 × 106 Gy) préalable de cette céramique sur sa vitesse de relâchement du césium en milieu aqueux n'a été observé, ce qui va également dans le sens d'une bonne tenue à l'irradiation de cette matrice. Par ailleurs, des essais de lixiviation (tests dynamiques à 100 °C en mode soxlhet [15]) que nous avons réalisés sur un échantillon de hollandite au césium (BaCs0,28Fe0,82Al1,46Ti5,72O16) non irradié et irradié aux électrons (énergie :1,5 MeV, fluence : 1,2 × 1019 cm−2) vont également dans ce sens car aucun effet significatif de l'irradiation sur la capacité de la céramique à retenir le césium n'a été observé [10].
Remerciements
Nous remercions vivement le CEA et le GdR Nomade (GdR 2023, CNRS/Universités/CEA/AREVA/EDF) pour leur soutien financier concernant nos recherches sur les matrices de confinement spécifique pour radionucléides à vie longue. Nous tenons aussi à remercier J-M. Costantini (CEA, Saclay) pour ses conseils et discussions concernant l'effet des irradiations électroniques sur la hollandite.