

1 Introduction
The increase in crude oil prices and desire to limit carbon dioxide emissions from fossil fuels implies the diversification of the sources of liquid fuels. In this context, the European Union has set an objective of substituting 20% of the conventional fuels with alternative fuels in the road transport sector by the year 2020 [1]. One possibility is to produce oil by pyrolysis of biomass such as wood waste, which is a renewable source. However, wood-derived bio-oil fractions resulting from this pyrolysis contain very important amounts (up to 45 wt%) of oxygenated compounds [2,3]. These molecules present multiple functions like aldehyde, ketone, acids, and alcohols. High oxygen content leads to deleterious properties such as high viscosity, thermal and chemical instability [3,4]. Therefore, upgrading of bio-oils by reducing their oxygen content is required to obtain a usable fuel. Hydrodeoxygenation (HDO), which occurs during hydroprocessing, refers to the high temperature hydrogen treatment of the feed to obtain oxygen-free molecules. Oxygen is then removed in the form of water. Sulfided hydrotreating catalysts such as CoMo/Al2O3 and NiMo/Al2O3 are commonly used in HDO [3–8]. These catalysts, which are massively used in hydrodesulfurization (HDS) and hydrodenitrogenation (HDN) processes have been investigated extensively [5,9–15].
It is now well accepted that active sites are located on the edges of MoS2 nanocrystallites that correspond to the (100) edge plane of the layered structure [5]. This crystallographic (100) plane exhibits alternative rows of sulfur-terminated layers (hereafter called S, or sulfur edge) and molybdenum-terminated layers (hereafter called M, or molybdenum edge) [16,17]. Stability of the catalysts is crucial, as interaction of water and oxygen-containing compounds with the catalysts can modify the sulfide structure of the catalyst edges [3]. Unfortunately, the stability of sulfide phases in presence of water, under usual HDO conditions, is still unknown.
Furan, one of the simplest oxygenated molecules present in HDO fractions, has been used to evaluate the activity of different catalysts. The reaction products of furan hydrodeoxygenation are butane and different isomers of butene, but the question of the primary products (tetrahydrofuran or butadiene) is still a matter of controversy; some mechanisms have been proposed in the literature [3,6] but they remain unclear. Density functional theory (DFT) calculations may provide insight into the furan HDO mechanism but require a detailed study of the adsorption of this molecule on the active MoS2 phase. A similar work concerning the thiophene adsorption [18], a typical model compound used in HDS studies, showed the importance of an accurate description of the surface taking into account the nature of the surrounding atmosphere [12–15,18]. Therefore, in a first step, the stable surfaces of the catalyst MoS2 under usual HDO conditions are determined using periodic DFT calculations. Then furan adsorption geometries on the MoS2 (100) surfaces are systematically investigated on surfaces stable under HDO catalytic conditions.
2 Computational methods
The density functional theory (DFT) calculations were performed with the Vienna Ab initio Simulation Package (VASP) [19,20] based on Mermin's finite temperature DFT [21]. The wavefunction is expanded in a plane wave basis set and the electron–ion interactions are described using the projector augmented plane wave (PAW) method [22,23]. The solution of the Kohn–Sham equations was improved self-consistently until a difference lower than 10−5 eV was obtained between successive iterations. The calculations were performed at the Γ point with a cut-off energy of 450 eV and a Methfessel and Paxton [24] smearing with σ = 0.1 eV. The exchange correlation functional of Perdew and Zunger [25] was used with the generalized gradient corrections proposed by Perdew et al. [26]. For more details about the code, we refer the reader to Ref. [19,20].
Throughout this work, we used the large super cell (12.641 × 12.294 × 20.000 Å3) shown in Fig. 1, which contains four elementary MoS2 units in the x direction, four in the z direction, and two layers along the y-axis (Fig. 1). Previous studies [12–15,17,27,28] showed that this model is suitable to predict the electronic and structural properties of the MoS2 surface. A vacuum layer of 10 Å is located above the MoS2 slab in the z direction in order to avoid interactions between slabs. The two upper rows were allowed to relax until forces acting on ions are smaller than 3.10−2 eV Å−1. The two lower were kept fixed to simulate bulk constraints.
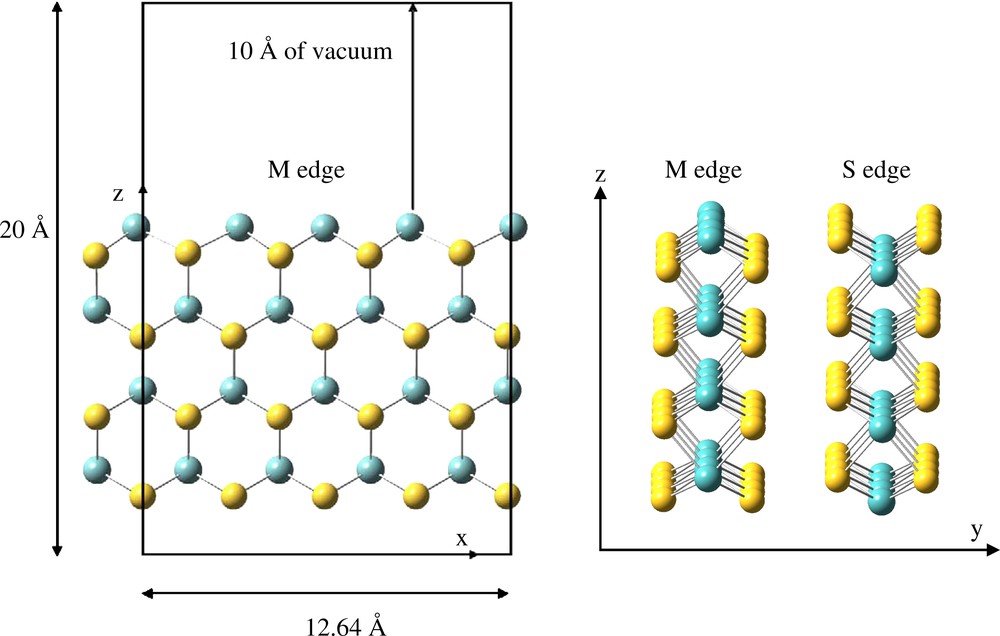
Cell showing the perfect MoS2 (100) surface. Molybdenum atoms are in blue, sulfur atoms are in yellow.
3 Surface stability in the presence of water
3.1 Reference surface
Under working conditions, the presence of H2, H2S and H2O will modify the stoichiometry and the nature of the atoms on the edges of the catalyst active phase. Experimentally, the catalyst is first sulfided and kept under hydrogen pressure before the introduction of the feed containing oxygen compounds. Therefore the initial structure is only dependent on the H2/H2S partial pressure ratio. In reducing conditions, the most stable surface [27,28], which will be used as reference, is presented in Fig. 2. We then investigate sulfur–oxygen exchange reaction and various oxygen stoichiometries in order to determine the effect of water on the surface stability.
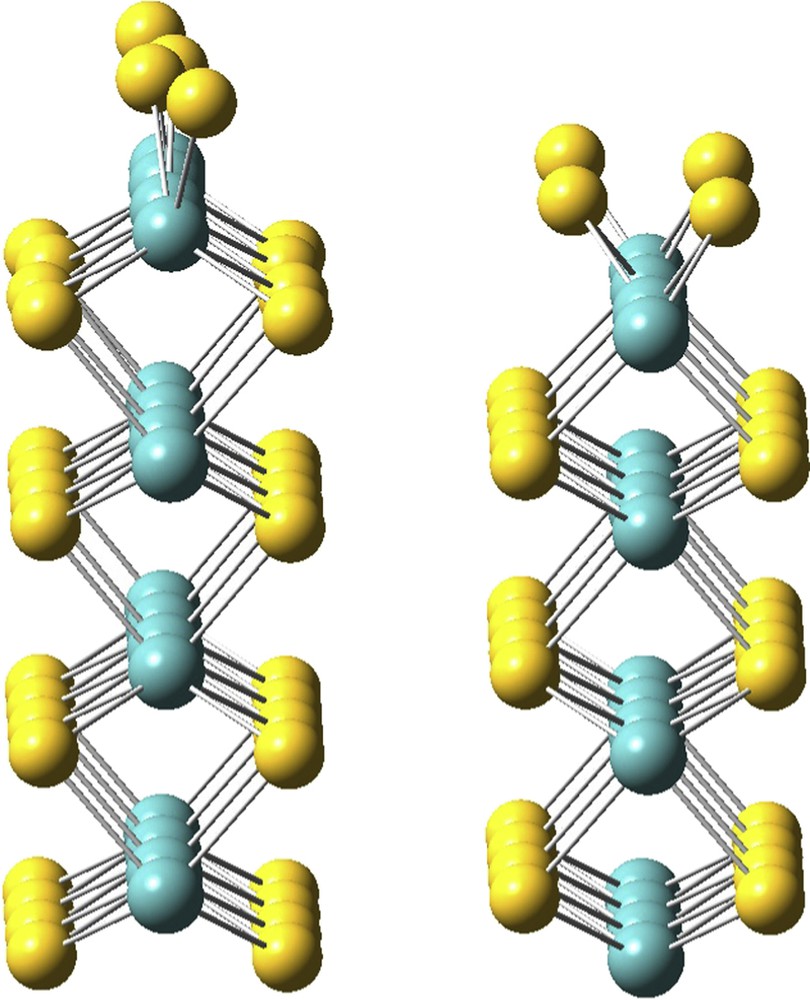
MoS2 stable surface when 0.05 < H2S/H2 < 10,000 [23,24] that will be used as reference. The M-edge is on the left, the S-edge is on the right.
Under hydrotreating conditions, the molybdenum edge is covered with sulfur atoms (Fig. 2), yielding six-fold-coordinated Mo edge atoms [15,27,29]. The surface undergoes an important reconstruction with the formation of short (2.8 Å) and long (3.5 Å) Mo–Mo distances. Within the same partial pressure and temperature range, the sulfur edge is reduced: half of the sulfur atoms are removed from the surface leading to four-fold-coordinated molybdenum atoms.
3.2 Sulfur–oxygen exchange
Under hydrodeoxygenation conditions, a large amount of water is produced because of the very high content of oxygenated compounds in the feedstock [3]. These compounds and H2O can depose oxygen atoms on the unsaturated molybdenum atoms and modify the surface state. Sulfur–oxygen exchanges can take place on the stable reference surface named hereafter surface [0-0] according to reaction (1).
| (1) |
| x + y = n |
Taking water as the oxygen source, successive exchanges (up to n = 6) were studied on both edges. The calculated surfaces will be noted [Z − y] or [x − Z] to indicate that the treated edge carries x or y oxygen atoms, while the second one is undefined as we assumed that the interaction between the 2 edges is small and that the edges can be studied separately. The computed electronic energies of each surface are reported in Table 1. Whatever the edge and the number of oxygen atom, the reaction is endothermic but the exchange will be favoured on the sulfur edge. Indeed, the energy required per S–O exchange for n = 1–4 is around 0.9 eV on the M-edge whereas it is around 0.4 eV on the S-edge (Table 1). On the metallic edge, the exchange energy is similar for the atom on the edge and for the neighboring atoms on the basal plane, and thus the exchange energy is almost constant for the substitution of the first 4 atoms. On the sulfur edge, the exchange energy for the substitution of one edge atom is 0.4 eV (for 1–4 exchange) while it is 0.9 eV for the substitution of the neighboring basal plane atoms. The average exchange energy will increase when all the 4 sulfur atoms on the edge have been substitute by oxygen ones.
Electronic contribution for the S–O exchange reaction from the reference surface [0-0].
| Surface | n | Energy required to perform the nth exchange (eV) | ΔEn (eV) | Energy per exchanged oxygen atom ΔEn/n (eV) | |
| M-edge | [Z-1] | 1 | 0.90 | 0.90 | 0.90 |
| [Z-2] | 2 | 0.75 | 1.65 | 0.83 | |
| [Z-3] | 3 | 0.97 | 2.62 | 0.87 | |
| [Z-4] | 4 | 0.91 | 3.53 | 0.88 | |
| S-edge | [1-Z] | 1 | 0.27 | 0.27 | 0.27 |
| [2-Z] | 2 | 0.39 | 0.66 | 0.33 | |
| [3-Z] | 3 | 0.50 | 1.16 | 0.39 | |
| [4-Z] | 4 | 0.46 | 1.62 | 0.41 | |
| [5-Z] | 5 | 0.85 | 2.47 | 0.49 | |
| [6-Z] | 6 | 0.82 | 3.29 | 0.55 |
3.3 Thermodynamic contributions
In order to define the most stable surfaces in the catalytic conditions, the S–O exchange reactions' Gibbs free energies are computed according to reaction (1):
where ΔEn = E(surface [x − y]) − E(surface [0-0])+ nE(H2S) − nE(H2O) is the electronic contribution for the S–O exchange reaction, and is the difference between hydrogen sulfide and water chemical potentials.
is computed with the partition functions of each molecule using the standard formulas of statistical thermodynamics [30,31]. ΔZPE refers to zero point energy (due to the vibrations) between the molecules.
This equation leads to a linear relationship between ΔrG and that is represented for each surface in Fig. 3 (M-edge) and Fig. 4 (S-edge). The calculations were performed at T = 350 °C, which corresponds to the working temperature of the catalyst and H2S/H2O partial pressure ratios between 10−4 and 100 (0.0001 < H2S/H2O < 100). The most stable surface is obviously that presenting the lowest free energy that is represented on the diagrams by the lowest line for each P(H2S)/P(H2O) partial pressure ratio.
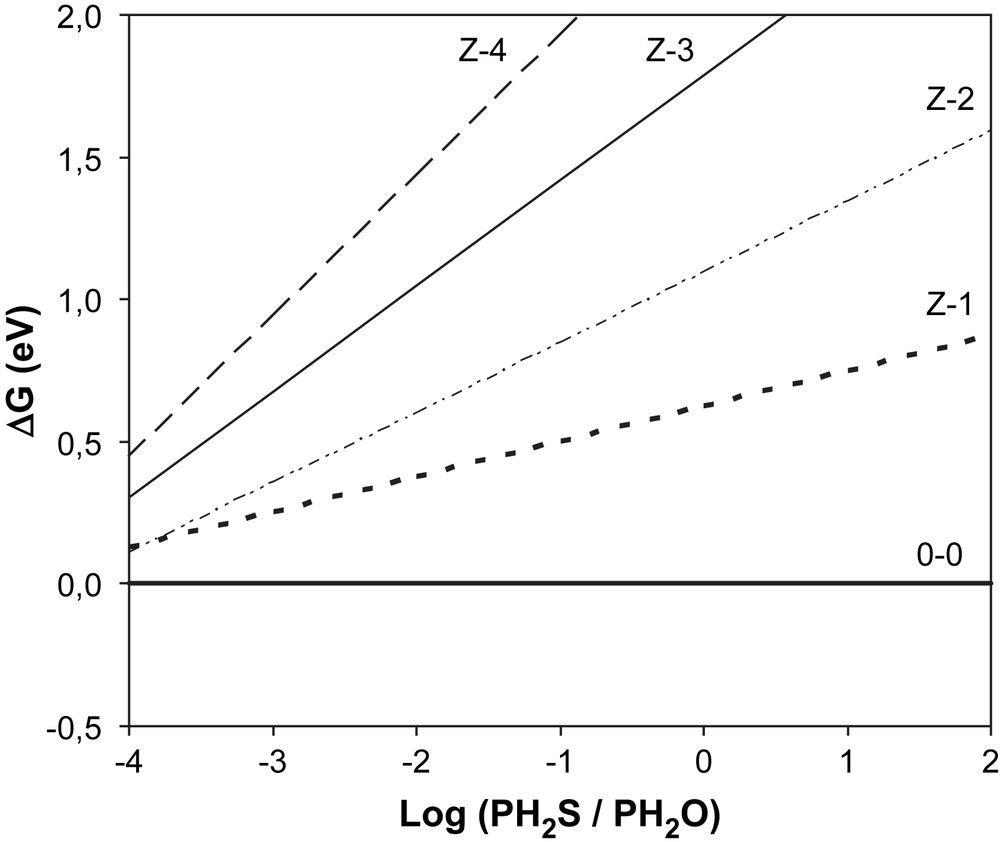
Metallic edge: variation of the Gibbs free energy versus .
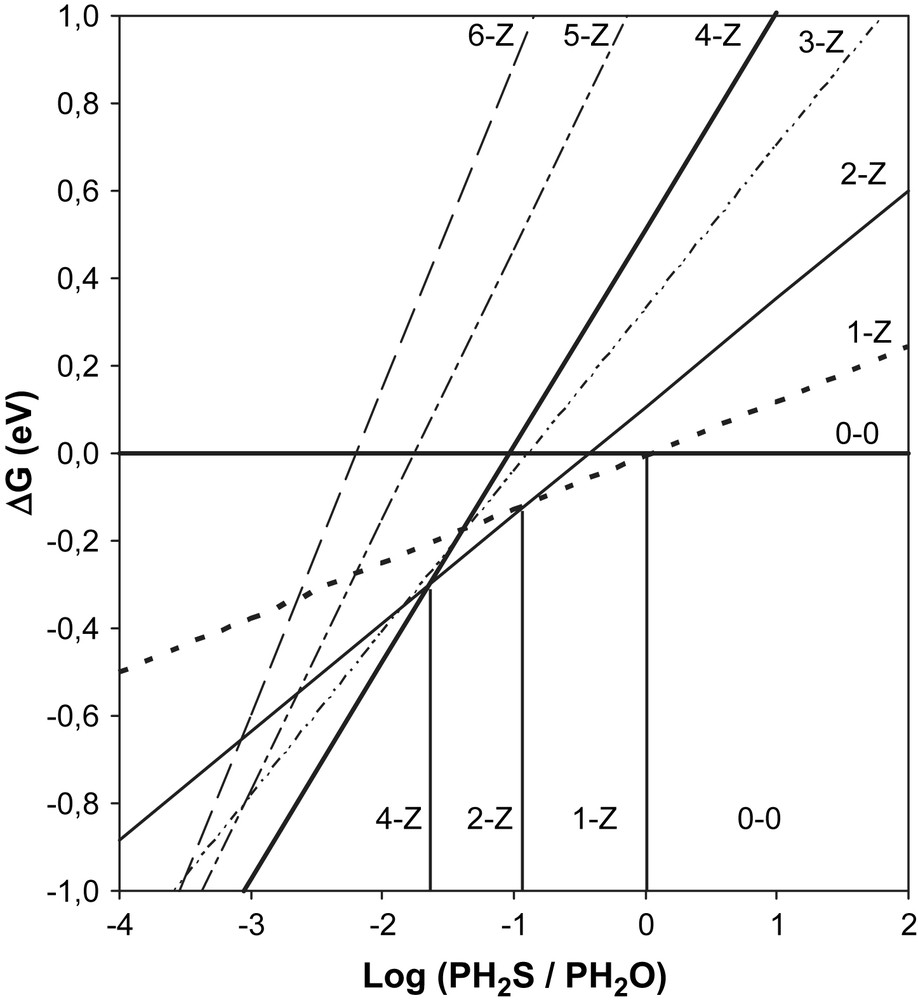
Sulfur edge: variation of the Gibbs free energy versus.
For the M-edge, no [Z − y] surfaces are stable in the pressure range we considered. We can thus conclude that S–O exchange does not occur on the M-edge and that the metallic edge is stable within the entire domain studied. On the other hand, the surface of the S-edge can be partially oxygenated [1-Z] if H2S/H2O < 1 (Fig. 5). The surface will be fully oxygenated [4-Z] for H2S/H2O < 0.025, which means H2O/H2S > 40 (Fig. 6). These results show that it is essential to keep a partial pressure in H2S during the HDO process in order to avoid the desulfurization of the catalyst and to maintain the catalytic activity [32].
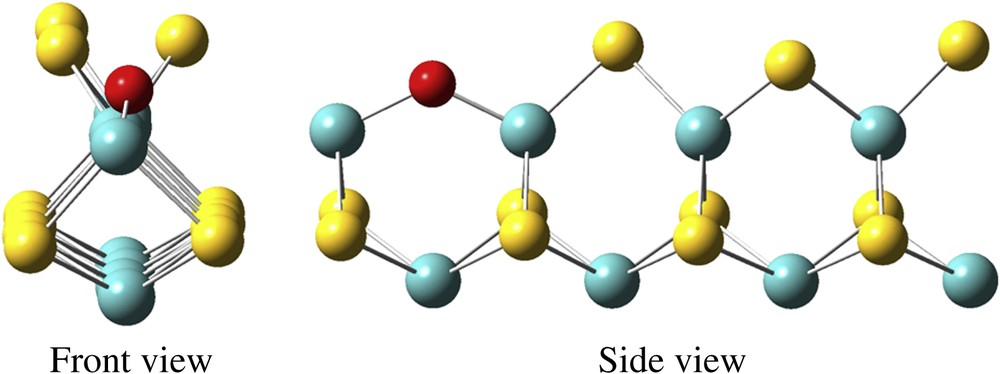
S-edge of [1-Z] surface: stable when 0.01 < H2S/H2O < 1. Molybdenum atoms are in blue, sulfur atoms are in yellow, oxygen atoms are in red.
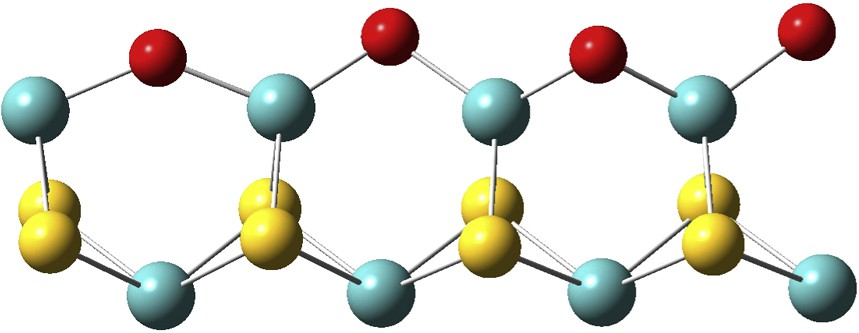
S-edge of [4-Z] surface: stable when H2S/H2O < 0.025, which means high water partial pressure.
4 Furan adsorption
The study of furan adsorption has been divided in three parts. The first part deals with the adsorption on the metallic edge, the second part concerns the non-exchanged sulfur edge, while the third part studies the adsorption on the fully oxygenated sulfur edge i.e. the [4-Z] surface. The adsorption on the stable surface and on coordinatively unsaturated sites (CUS), created by reaction with hydrogen under reductive conditions, was considered. All through this work, a positive adsorption energy corresponds to an exothermic process.
4.1 Adsorption on the metallic edge
Whatever the tested starting geometries, the furan does not adsorb on the metallic edge. The molecule is always desorbed, without any activation barrier, during the geometry optimization.
It is generally assumed that HDS, HDN and HDO reactions proceed on the CUS of the MoS2 edges. Hence, we also considered the adsorption on vacancies generated by removing one sulfur atom from the edge stable under reducing conditions. This vacancy formation is an endothermic reaction. On the metallic edge, because of the surface reconstruction [15,27,29], two types of vacancies can be created (Fig. 7), yielding two five-fold-coordinated Mo atoms (Mo5c). The most stable defective surface exhibits a “long” vacancy (denoted by L hereinafter) created by removing a sulfur atom originally above the long Mo–Mo distance, initially 3.5 Å and 3.6 Å after optimization (ΔrE = 0.54 eV). The formation of a “short” vacancy (denoted by S hereinafter) between the two closer Mo atoms (initially 2.8 Å and 3,2 Å after optimization) is 0.25 eV more endothermic (ΔrE = 0.79 eV). The η1 adsorption is possible on both vacancies (Fig. 8), with the oxygen atom of the molecule located in a bridging position between the two Mo5c atoms, and leads to the same geometry. The adsorption energy depends on the type of vacancy: its energy is 0.04 eV for the L vacancy and 0.29 eV for the S vacancy. The global process is endothermic and needs the same energy (0.50 eV) on both vacancies as the final geometries are the same.
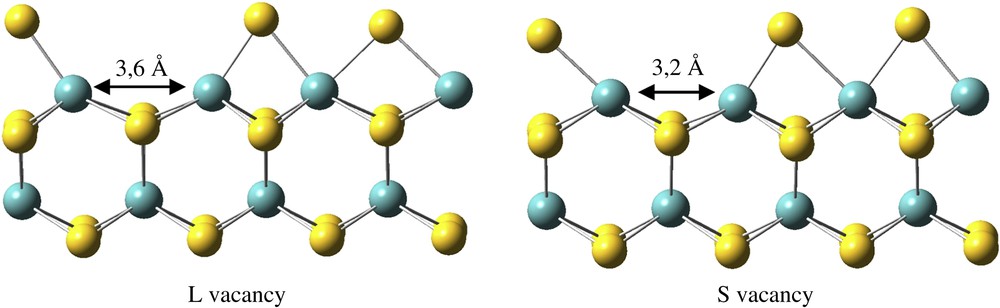
L and S vacancies located on the M-edge.
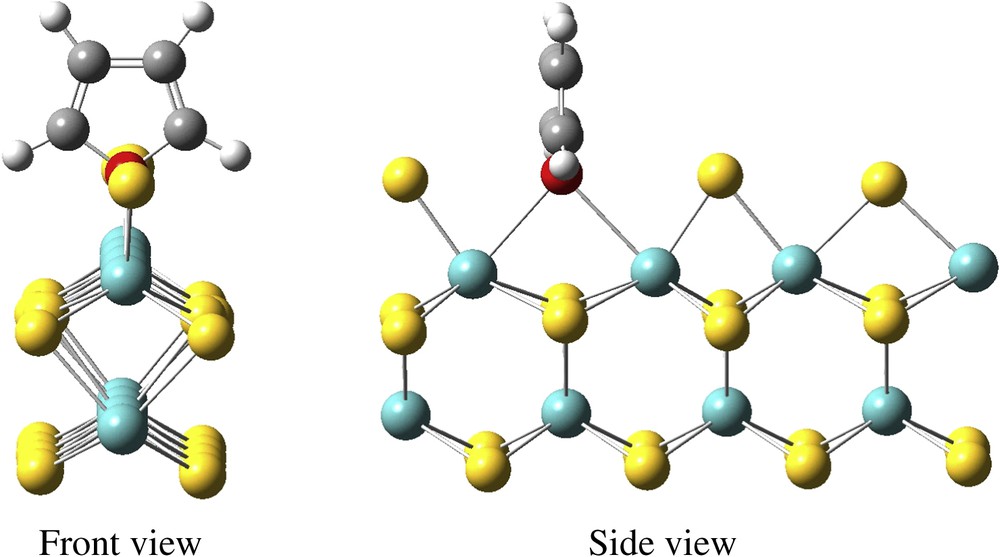
Furan adsorbed (η1) on an L vacancy located on the M-edge.
To check the affinity of the Mo5c for an oxygen atom that already forms two bonds, we computed the H2O adsorption energies on these two vacancies. We also calculated the H2S adsorption energies in order to evaluate the competition effect. The results, reported in Table 2, show that the adsorptions of H2O and H2S on both vacancies compete with furan adsorption. Concerning the L vacancy, the adsorption energy of H2O is higher than the H2S one. In contrast, these energies are similar on the S vacancy. These findings may be related to the Mo–SH2 bond (2.55 Å) length which is slightly larger than the Mo–OH2 one (2.50 Å).
Adsorption energies (eV) for different molecules on CUS located on the M-edge.
| Vacancy | Furan | H2O | H2S | Thiophene |
| L | 0.04 | 0.47 | 0.24 | 0.15 |
| S | 0.29 | 0.10 | 0.07 | −0.04 |
A flat adsorption on the metallic edge with one vacancy is possible (Fig. 9). This η5 adsorption is endothermic (Eads = −0.85 eV) on the L vacancy and requires an important surface reconstruction; one S atom is displaced and located on top of one Mo atom, forming one S–S bond with its neighbor. The energetic cost of this reconstruction is very high (1.83 eV), hence this kind of adsorption should not occur.
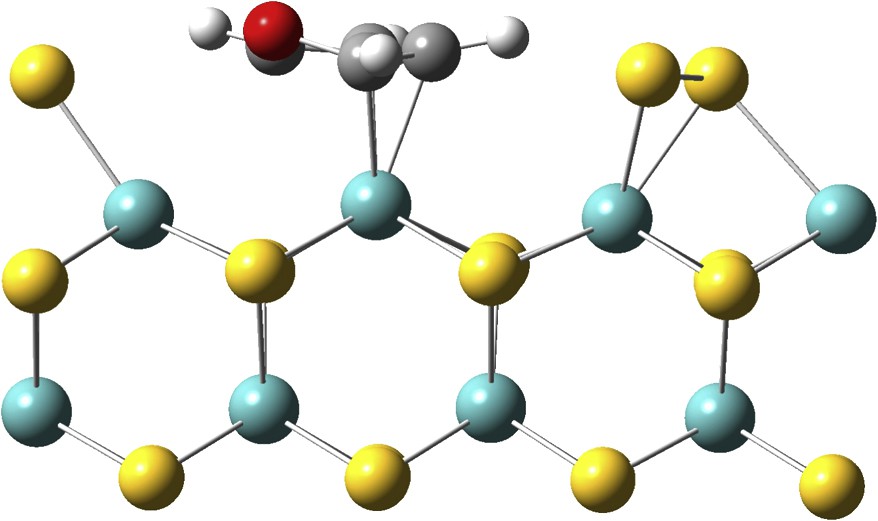
Furan adsorbed (η5) on an L vacancy located on the M-edge.
4.2 Adsorption on the reference sulfur edge
Whatever the starting geometries, the furan does not adsorb on the sulfur edge. This is quite surprising since thiophene can adsorb directly on this edge (Table 3) [18]. The average molybdenum–oxygen distance is shorter than the molybdenum–sulfur one. The distance between the furan and the edge would be shorter than the thiophene one, which increase the steric repulsion between the molecule and the edge. The adsorption on the stable edge is not possible and therefore the creation of a CUS is required for the adsorption of furan on this edge.
Adsorption energies (eV) for different molecules on the stable S-edge.
| Surface | H2S/H2O | Furan | Thiophene |
| [0-0] | >1 | No adsorption | 0.18 |
| [4-Z] | <0.025 | No adsorption | No adsorption |
The vacancy formation by removal of one S atom is more endothermic (ΔrE = 1.61 eV) on the sulfur edge than on the molybdenum edge. This value is so high that the creation of such CUS should be very limited during the catalytic reaction. On this vacancy, the adsorption energy is slightly exothermic (Eads = 0.26 eV). Flat adsorptions are not possible on this edge.
4.3 Adsorption on the fully oxygenated sulfur edge
Whatever the starting geometry, no minimum has been found for furan or thiophene adsorbed on the fully oxygenated sulfur edge. Once again, a CUS would be required to allow adsorption of molecules on this edge. The creation of such a CUS is even more endothermic (ΔrE = 1.98 eV) on this edge than on the reference sulfur edge. This value is so high that the creation of such CUS should not occur during the catalytic reaction. We thus assume that this kind of edge cannot be reactive for the furan HDO.
4.4 Discussion
To compare the adsorption on the defective and stable surfaces, the whole process must be considered. The adsorption on defective surfaces must take into account the vacancy formation according to reaction (2), while reaction (3) describes the adsorption of furan on the stable surfaces:
| Surface–S + H2(g) + OC4H4(g) = Surface–OC4H4 + H2S(g) | (2) |
| Stable surface + C4H4O(g) = Surface–OC4H4 | (3) |
The energies of the various reactions involved in the adsorption on the two types of surfaces are summarized in Table 4. Under reductive conditions corresponding to the reaction conditions, the most probable adsorption mode is η1 on a CUS located on the M-edge. These results demonstrate that CUS are required for furan adsorption on the edges of the MoS2 nanoparticles and that the complete adsorption process (vacancy formation plus adsorption) is endothermic. These results are in agreement with experimental data [33] on MoS2 using radioactive sulfur. The introduction of dibenzofuran in the reactor leads to the departure of sulfur atoms, which is a strong evidence of vacancy formation.
Computed reaction energies (eV) for furan and thiophene adsorption starting from the stable surfaces under reductive conditions.
| Edge/surface | Reaction | Adsorption mode | Furan | Thiophene |
| M/stable surface | (2) | No adsorption | ||
| M/defective surface | (3) | η1 | 0.50 | 0.38 |
| M/defective surface | (3) | η5 | 1.39 | 1.08 |
| S/stable surface | (2) | η1 | No adsorption | 0.18 |
| S/defective surface | (3) | η1 | 1.35 | 0.73 |
On the other hand, thiophene can adsorb on the non-defective sulfur edge. Therefore, the furan deoxygenation reaction mechanism will be quite different from the thiophene desulfurization and the correlation of thiophene HDS activity with HDO one will not be straightforward.
5 Conclusion
In this work we showed that the stability of the edges of the MoS2 nanocrystallites depends on the H2S/H2O pressure ratio and that the introduction of H2S in the feedstock is mandatory in order to avoid oxygenation of the catalyst. Furan adsorption on MoS2 surfaces that is stable under HDO conditions is always endothermic. On defective surfaces, the adsorption is exothermic, but vacancy creation is endothermic and this reaction is activated. The η1 adsorption on a CUS of the metallic edge is the most favorable adsorption mode and is probably the first step of the reaction mechanism.
Acknowledgements
This work has been performed within the ANR funded Programme national de recherche sur les bioénergies-ECOHDOC, a joint project of the Centre national de la recherche scientifique (CNRS), the universities of Caen, Lille and Poitiers, and TOTAL. The authors thank the USTL Centre de ressources informatiques (partially funded by FEDER) for allocating CPU time.