

1 Introduction
A great variety of molecular cages have been assembled by complexation of polypyridinyl ligands with the metal cation fragments [Pd(bipy)]2+ and [Pd (en)]2+ under thermodynamic control, thanks to the lability of non chelating Pd–N bonds in polar solvents [1]. The resulting species can encapsulate guests, which may also play the role of templates [1b,c], and provide a confined environment able to control the regio- and stereoselectivity of various reactions [1d,e] or stabilize otherwise elusive species [1f]. In general, the self-assembled molecular cages form geometrical structures whose sides and vertices (or edges) are occupied by the polypyridinyl ligands and the metal-complex fragments, respectively. Very few examples however, feature systems in which the latter are used to fix the shape of elaborated polytopic ligands [2].
The pyrimidinyl coordinating subunit has been used for the construction of several supramolecular cage structures [3], in particular those incorporating the hypothetical metallacalix[3]arene fragment that would result from cyclotrimerisation of the pyrimidine-Pd2+ coordination motif [3a,b,f]. We have synthesized two tris (pyrazol-1-ylmethyl) benzene tripods (1 and 2), whose pyrazolyl position 3 is substituted by a 5-pyrimidinyl fragment (Scheme 1). It was expected that bridging of the pyrimidinyl nitrogen atoms with [Pd (bipy)]2+ would form macrotricycles that are metallo-organic analogues of the reported tris (pyrazole)-incorporating organic macrotricycles, which are able to encapsulate NH4+, mainly by hydrogen bonding interactions (Scheme S1) [4]. This work shows that, instead of the anticipated [Pd3(bipy)3(1)]2+ and [Pd3(bipy)3(2)]6+ macrotricycles, the simple metallamacrocyclic structures [Pd(1)(bipy)]2+ and [Pd(2)(bipy)]2+ are obtained, indicating that the incorporation of additional [Pd(bipy)]2+ complex fragments is unfavorable. The metallamacrocycles display remarkable dynamic properties at the 1H NMR timescale: whereas at room temperature they have the expected C2v symmetry, at high temperature (423 K) they show an average C3v symmetry, because the [Pd(bipy)]2+ fragment hops between the pyrimidinyl coordinating units. As ΔGc‡ ≈ ΔHc‡ for this process, the activation enthalpy for the dissociation of the Pd–N bond could be estimated to ≈ 70 kJ mol–1.
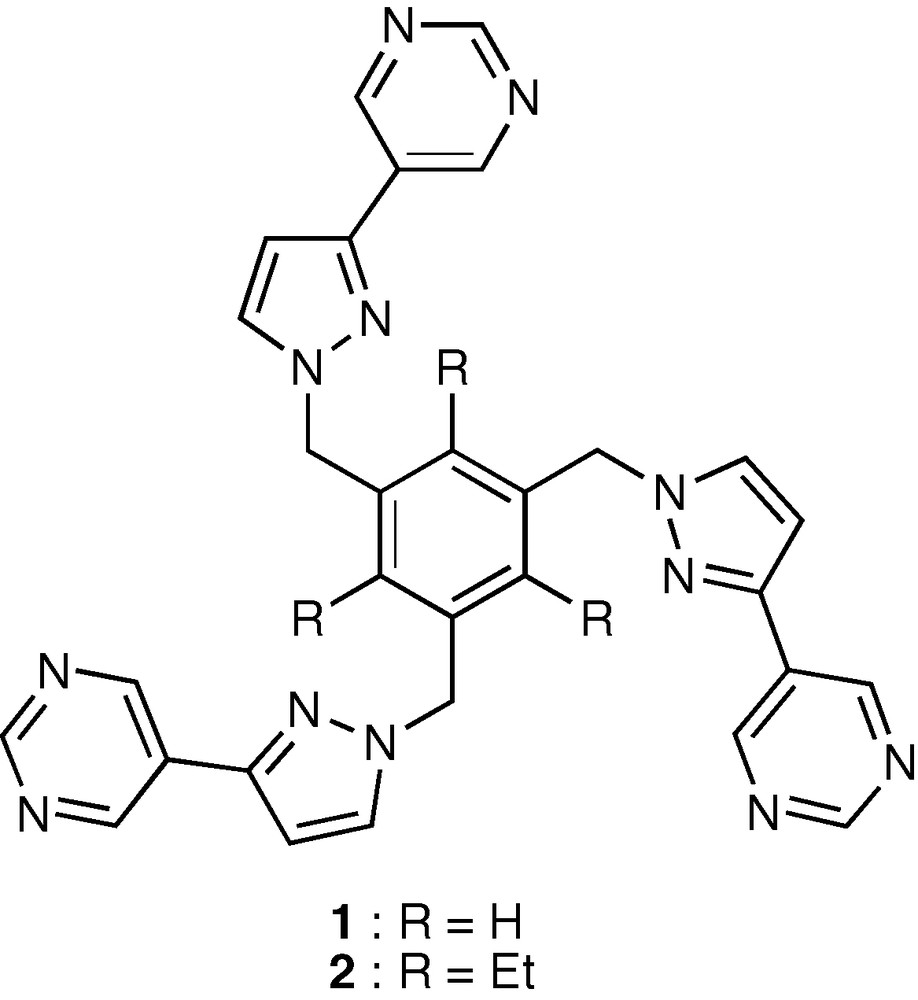
Structural formulae of tripod ligands 1 and 2.
2 Results and Discussion
The direct precursor of 1 and 2, namely 5-(1H-pyrazol-3-yl)-pyrimidine (5) was synthesized using a classical preparation of pyrazoles, that is condensation of the appropriate β-N,N-dimethylaminoenone with hydrazine (Scheme 2) [5]. Thus 3-(dimethylamino)-1-(5-pyrimidinyl)-2-propen-1-one (4), readily obtained (93% yield) by heating 1-(5-pyrimidinyl)-2-ethanone (3) [6] in dimethylformamide dimethylacetal, was reacted with hydrazine hydrate in the presence of acetic acid, affording 5-(1H-pyrazol-3-yl)-pyrimidine 5 in 92% yield after purification. Noticeably, a very efficient preparation of this compound (four steps, 64% overall yield from 5-bromopyrimidine) has been recently reported. It involves the Suzuki cross-coupling reaction between 5-bromopyrimidine and 1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazole in 92% yield, followed by quantitative hydrolytic cleavage of the THP protection [7].
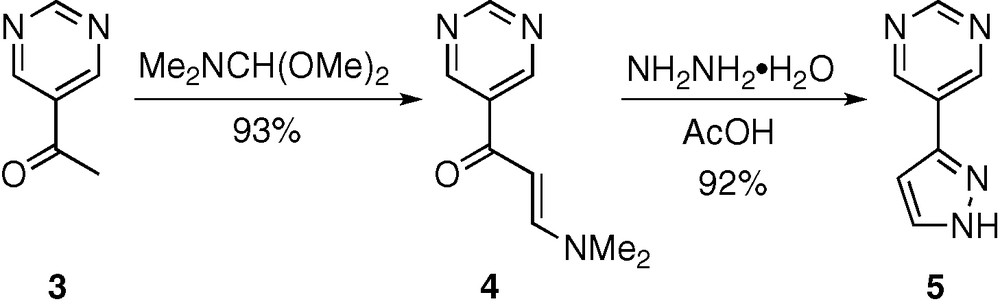
Preparation of 5 starting from 3.
For the preparation of the target tripod compounds 1 and 2, pyrazole 5 was deprotonated first [8], and the resulting pyrazolate reacted with the appropriate tris-electrophile, respectively 1,3,5-tribromomethylbenzene [9] and 1,3,5-tribromomethyl-2,4,6-triethylbenzene [10]. The corresponding tripods were obtained in 53% (1) and 79% (2) yields after chromatographic purification. Both have 1H NMR spectra in agreement with their expected D3h symmetrical structure.
In the initial complexation experiments, three equivalents of [Pd(bipy)(ONO2)2] [11] were added to a solution of tripod 1 in d6-dmso at room temperature1. The 1H NMR spectrum of the mixture showed the signals of starting [Pd(bipy)(ONO2)2] along with those of a species that has not the expected C3-symmetry, because two sets of signals in 2:1 ratio can be identified for the tripod protons. The same product is formed quantitatively upon using only one equivalent of [Pd(bipy)(ONO2)2]. Complete analysis of the corresponding 1H and 13C NMR spectra using 1H/1H COSY and ROESY, 1H/13C HSQC and HMBC, indicates that the [Pd(bipy)]2+ fragment is complexed by two pyrimidinyl subunits of the tripod, thus closing a 20-membered chelate ring, and leaving the third pyrimidinyl arm pendant (Scheme 3). This is corroborated by the comparison of the 1H NMR spectra of 1 and [Pd(1)(bipy)]2+ (Table S1), which shows that the protons of the free pyrimidinyl arm have chemical shifts that are close to those of 1, whereas protons 2 and 6 ortho to the coordinated pyrimidinyl nitrogen are deshielded by metal coordination (Δδ = +1.00 ppm in the latter case). Significantly, intercomponent Nuclear Overhauser effect (NOE) correlations are found between these latter protons and the α protons of the bipy ancillary ligand. 2D 1H/1H EXSY of [Pd(2)(bipy)]2+ clearly shows that intramolecular exchange processes are taking place on the NMR timescale (Fig. 1). They exchange homologous protons of the coordinated and uncoordinated pyrimidinyl pendant arms (e.g. 4/4° or 4′/4′′), on the one hand, and homologous protons of the bound pyrimidinyl fragments (e.g., 4/6), on the other hand. Accordingly, VT 1H NMR spectra were recorded in the 293–423 K temperature range. This is illustrated for the case of [Pd(1)(bipy)]2+ in Fig. 2. As the temperature is increased, all the signals (major and minor) of the tripod protons broaden, giving rise to a sequence of coalescence phenomena (characterized by Δν in the slow exchange limit, and Tc, Table 1), and sharpen again to form a unique set at 423 K. These results can be interpreted as represented in Scheme 4. Whereas at room temperature the coordination of the [Pd(bipy)]2+ complex fragment to the tripod is fixed on the 1H NMR timescale, this is not the case at high temperature (423 K), where it hops from a pyrimidinyl site to the other. The fluxional process combines ligand and site exchange (resulting from rotation about the 5-C–3′-C bond), and involves Pd–N (pyrimidine) bond cleavage. The computed exchange rates kc and free activation energies at the coalescence (ΔGc‡) are collected in Table 1. Remarkably, ΔGc‡ = 68.1 kJ mol–1 does not depend on temperature, which indicates that ΔSc‡ = 0. This result proves that the “merry-go-round” process of [Pd(bipy)]2+ takes place intramolecularly, presumably by nucleophilic attack of the free pyrimidinyl arm on the bound Pd2+ cation, without complete decoordination of the former.
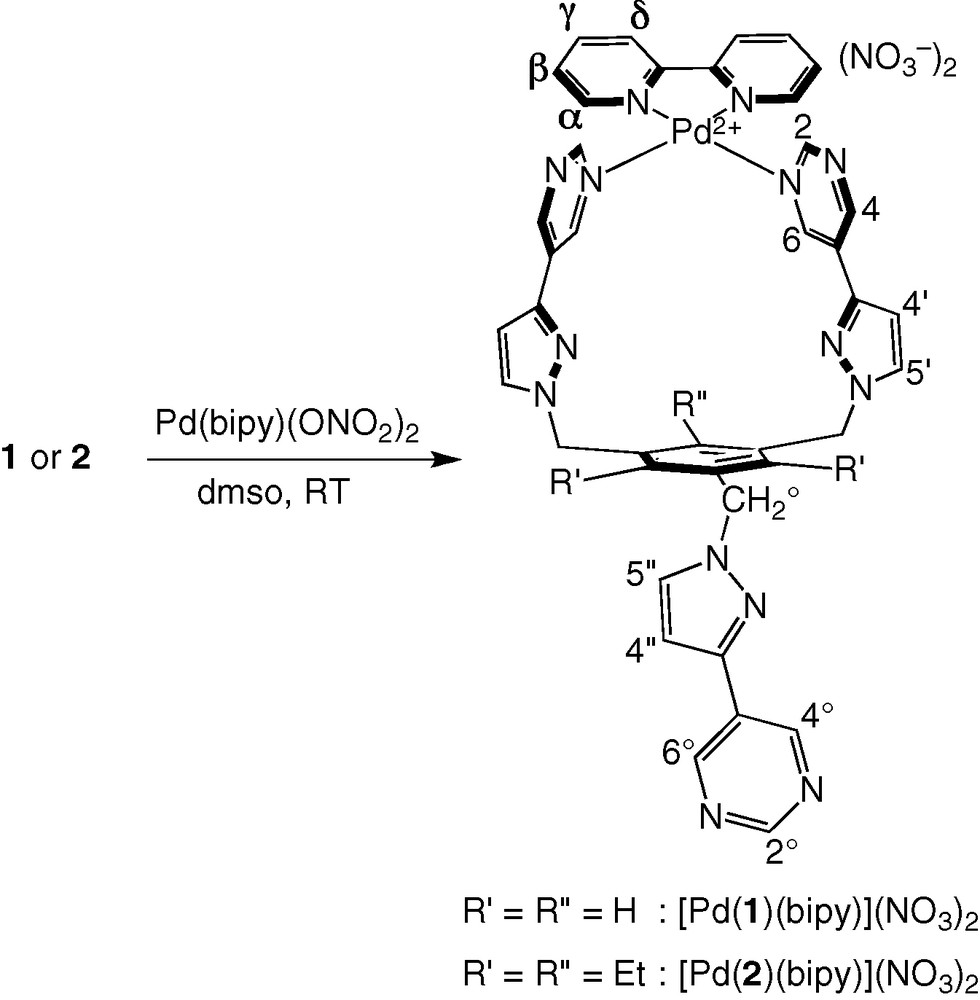
Formation of [Pd(1)(bipy)](NO3)2 and [Pd(2)(bipy)](NO3)2.
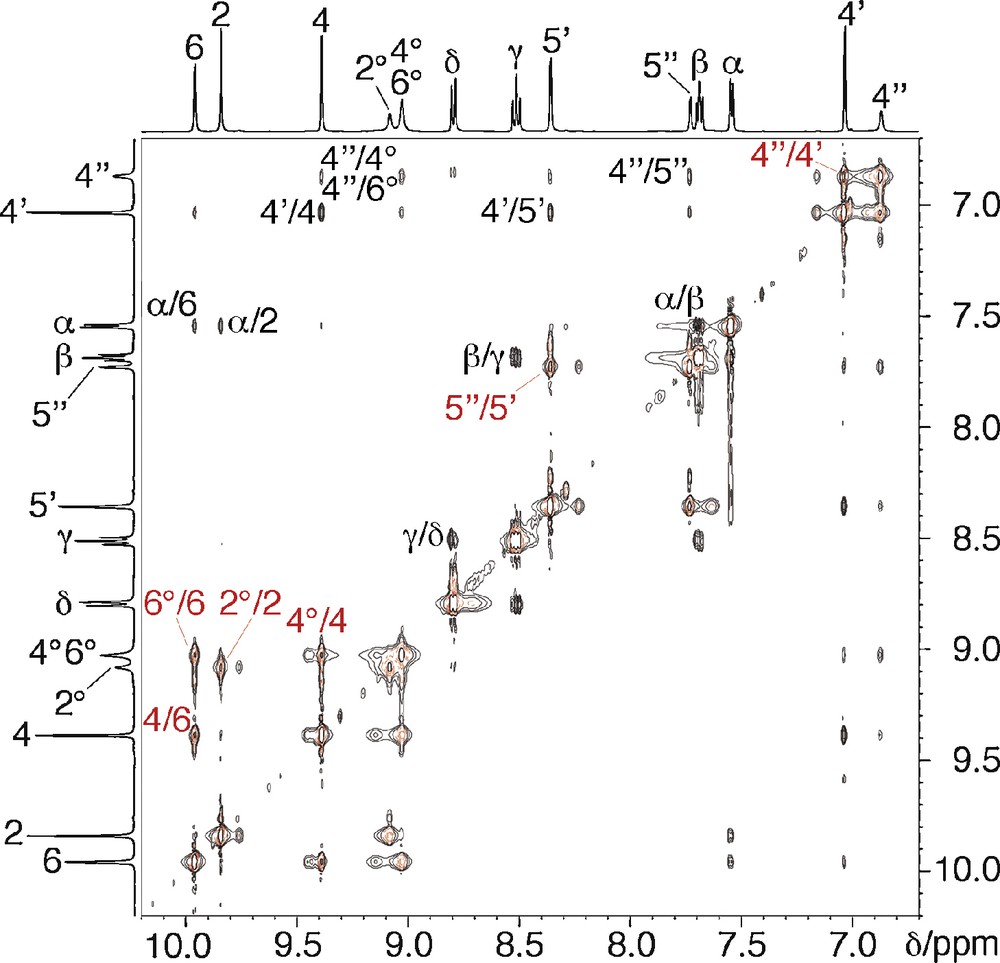
2D 1H/1H NMR EXSY spectrum (aromatic region) of [Pd(2)(bipy)]2+ in d6-dmso at 298 K (mixing time: 300 ms). Main NOE correlations and exchange spots are indicated in black and red, respectively.
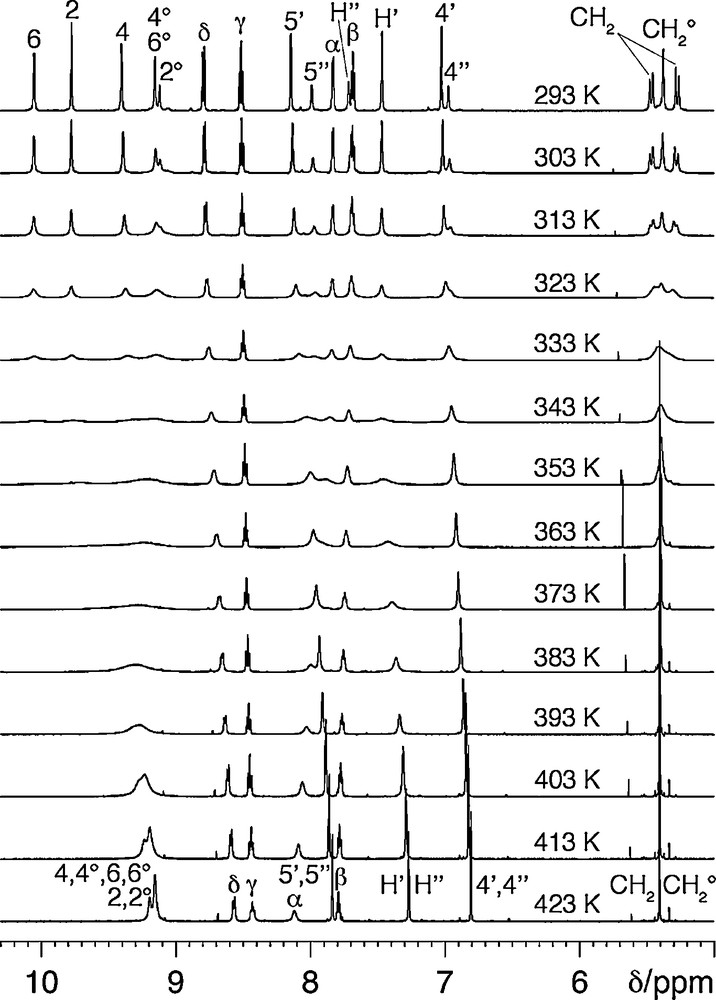
Sequence of variable temperature spectra of [Pd(1)(bipy)](NO3)2 in d6-dmso.
Data obtained from the VT 1H NMR spectra of [Pd(1)(bipy)]2+ and [Pd(2)(bipy)]2+.
| Compound | Probe protons | Tc/K | Δν/Hza | kc/s–1b | ΔGc‡ /kJ mol–1c,d |
| [Pd(1)(bipy)]2+ | 4′, 4′′ | 323 | 30 | 67 | 68 |
| 5′, 5′′ | 338 | 90 | 200 | 68 | |
| H′, H′′ | 343 | 150 | 333 | 68 | |
| 4, 4° | 343 | 150 | 333 | 68 | |
| 2, 2° | 358 | 395 | 877 | 68 | |
| 6, 6° | 363 | 540 | 1200 | 68 | |
| [Pd(2)(bipy)]2+ | 4′, 4′′ | 358 | 100 | 222 | 72 |
| CH2, CH2° | 358 | 120 | 267 | 72 | |
| CH3′, CH3′′ | 373 | 330 | 733 | 72 | |
| CH2′, CH2′′ | 383 | 715 | 1588 | 71 |
a Frequency difference at the slow exchange limit (293 K).
b kc (s–1) = (2–1/2)πΔν.
c ΔGc‡ (kJ mol–1) = 2.303 × 8.314 × Tc(10.319 + logTc–logkc).
d Estimated error: 1 kJ mol–1, assuming σ(Tc) = 5 K and σ(Δν) = 5 Hz.
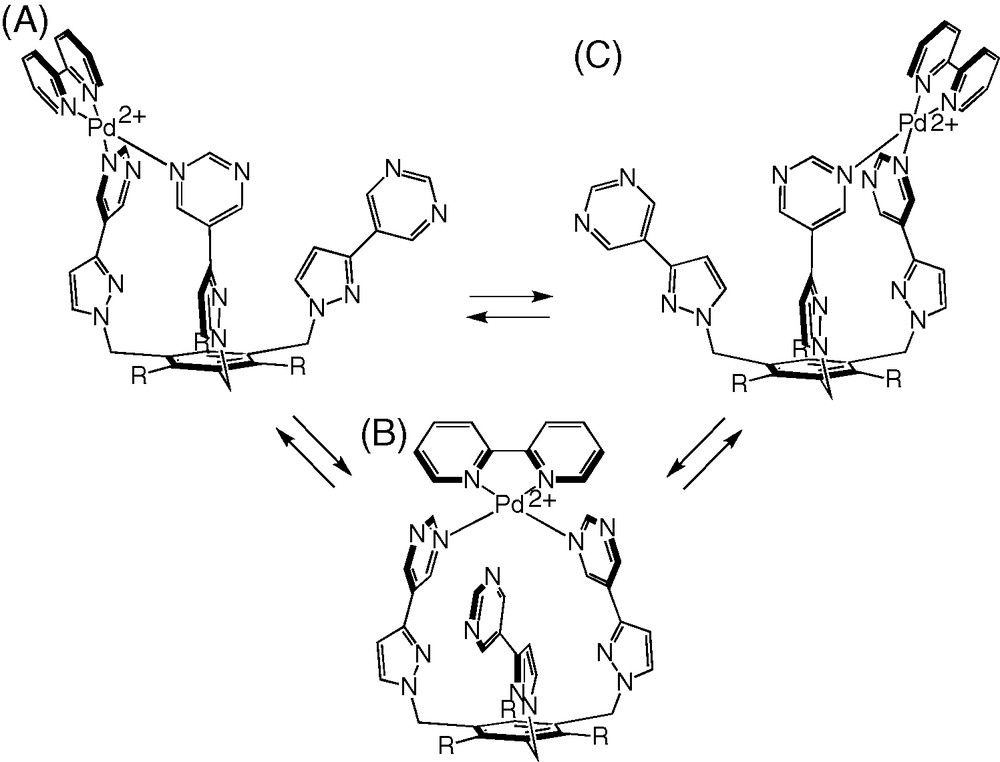
The “merry-go-round” process of the [Pd(bipy)]2+ metal-complex fragment.
The complex [Pd(2)(bipy)]2+ exhibits similar behavior, with a slightly higher ΔGc‡ value (71.5 kJ mol–1). This could arise from the increased rigidity of tripod 2 by comparison with 1, due to the ethyl substituents, which reduces the mobility of the uncomplexed arm. Fluxional processes involving Pd–N bond rupture have been previously observed in Pd complexes with terpyridines [12a], and 2-pyridyl [12b,c] or N-pyrazole [12d]-substituted triazines. For example, in the case of [Pd(p-CF3C6F4)2(tpt)], where tpt is 2,4,6-tris (2-pyridyl)-1,3,5-triazine, ΔG‡ = 72 kJ mol–1 for the 1,4 metallotropic shift (Scheme 5). The average ΔG‡ ≈ 70 kJ mol–1 value found for [Pd(1)(bipy)]2+ and [Pd(2)(bipy)]2+ is in keeping with that measured in the case of the tpt complex. In addition, our observation that ΔG‡ = ΔH‡ allows us to evaluate the enthalpy of activation for the dissociation of the Pd–N (pyrimidine) bond as ≈ 70 kJ mol–1.
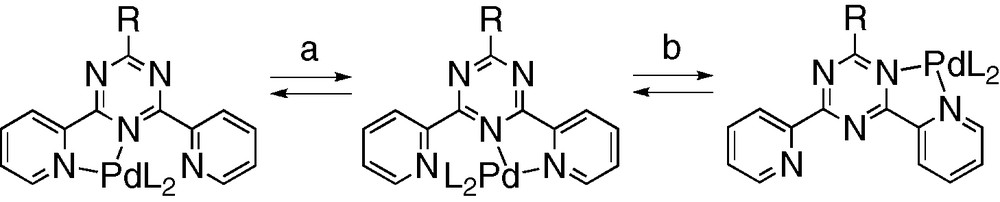
Fluxional processes occurring in [Pd(p-CF3C6F4)2(tpt)] (L = p-CF3C6F4, R = 2-pyridyl). (a) 1,4-Metallotropic shift. (b) Rotational hurdling process [12b,c].
Consideration of molecular models shows that the complexes [Pd(1)(bipy)]2+ and [Pd(2)(bipy)]2+ should exist under two different conformations, one in which the [Pd(bipy)]2+ metal-complex fragment hangs over the benzene platform and is located on the same side as the free pyrimidinyl arm (endo conformation), and the other in which the same fragment and the free pyrimidinyl arm lie on opposite sides (exo conformation), as illustrated in Scheme 6. Interconversion between these forms involves concerted rotation about the C (pyrazolyl)–C (pyrimidinyl) and Pd–N (pyrimidinyl) bonds or, in the case of [Pd(1)(bipy)]2+ only, rotation of the benzene ring about two C (benzene)–C (H2) bonds, which allows the free pyrimidinyl arm to pass through the metallo-organic macrocycle (Scheme 6). This latter process cannot obviously operate in the case of [Pd(2)(bipy)]2+ for steric reasons. Unfortunately, as only one species is observed at the lowest temperature possible in d6-dmso (293 K), these interconversion processes cannot be slowed down in this solvent, preventing one to experimentally demonstrate the possible coexistence of the endo and exo conformations.
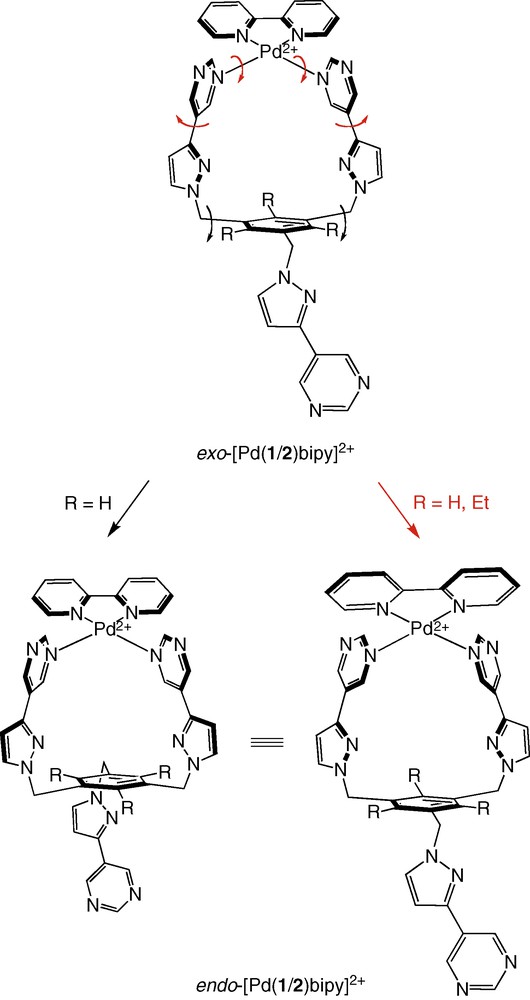
Mechanisms of interconversion between exo-[Pd(1/2)(bipy)]2+ and endo-[Pd(1/2)(bipy)]2+.
The [Pd(1/2)(bipy)]2+ complexes have been arbitrarily drawn in the exo conformation in Schemes 3 and 4. Nucleophilic attack of the pendant pyrimidinyl arm on exo-[Pd(1/2)(bipy)]2+ will produce necessarily endo-[Pd(1/2)(bipy)]2+. Therefore, exchange between any two of the three structures A, B, and C involves also the exo/endo interconversion process discussed above.
Finally, the inability of tripods 1 and 2 to take up three [Pd(bipy)]2+ complex subunits in dmso deserves being discussed. It could be due to either:
- • poor ligand preorganisation;
- • steric crowding of the peripheral bipy's;
- • lack of stabilization by dmso of the high charge density of the trinuclear species.
Whereas the first hypothesis cannot be ruled out, this is not the case of the second one, as replacing ancillary bipyridine by the smaller ethylenediamine leads to the same results. Solvent effects are more likely, as exemplified by the behavior of a C4-symmetric cavit-and bearing four pendant pyrimidinyl arms, which, upon pair wise connection with the same number of [Pd (en)]2+ complex fragments forms a metallo-organic macropolycycle [2b]. It must be noted that the desired, C4-symmetric tetranuclear complex was obtained in the less polar solvent mixture (methanol/water 5:1), whereas the reaction in water led to the incorporation of only two [Pd(en)]2+ moieties, producing a C2-symmetric system. By contrast, C3-symmetric metallo-organic cage molecules incorporating the metallacalix[3]arene [Pd(pyrimidinyl)]36+ motif were usually assembled in water [3a-c, 3g]. Whereas this latter solvent was not effective in promoting the formation of the trinuclear complexes because of solubility problems even after prolonged reaction times, future work will explore the effect of increasing the solvent polarity (dmso/water mixtures) or the ionic strength (added salt).
3 Conclusion
The lability of the Pd–N (aromatic) bond has been extensively exploited in thermodynamically-controlled self-assembly processes to produce elaborated macropolycyclic (cage) structures in water or polar solvents, such as dmso, acetone and MeOH. However, few reports have examined the dynamics of this important coordination bond. The data obtained in the present work are in agreement with those on related compounds, and permit to set the enthalpy of activation for the dissociation of this bond to ≈ 70 kJ mol–1.
4 Experimental section
4.1 General
THF was distilled from sodium/benzophenone. d6-dmso was dried on 3 Å molecular sieves. 1-(5-Pyrimidinyl)-1-ethanone [6], 1,3,5-tribromomethylbenzene [9], 1,3,5-tribromomethyl-2,4,6-triethylbenzene [10], and [Pd(bipy)(ONO2)2] [11] were synthesized as described in the literature. NMR spectra were recorded with Bruker 500 MHz Avance DRX or 600 MHz Avance II spectrometers.
4.2 3-(Dimethylamino)-1-(5-pyrimidinyl)-2-propen-1-one (4)
A solution of 1-(5-pyrimidinyl)-1-ethanone (0.324 g, 2.65 mmol) in dimethylformamide dimethyl acetal (5 mL, 37 mmol) was refluxed (100 °C) for 2.5 h. Excess of acetal was removed with a rotary evaporator. Sublimation of the residue (35 °C, 0.08 mbar) afforded 4 (0.436 g, 2.46 mmol) in 93% yield, as colored needles, mp 138-139 °C. Elemental analysis calcd (%) for C9H11N3O (177.05): C 61.05, H 6.26, N 23.73; found: C 61.34, H 6.50, N 23.70; 1H NMR (CDCl3, 500 MHz) δ 9.25 (s, 1 H, 2-H), 9.15 (s, 2 H, 4- and 6-H), 7.87 (d, 3JH,H = 12.1 Hz, 1 H, HCN), 5.61 (d, 3JH,H = 12.1 Hz, 1 H, HCO), 3.20 (s, 3 H, CH3), 2.97 (s, 3 H, CH3) ppm; 13C NMR (CDCl3, 126 MHz) δ 183.6 (CO), 159.7, 155.9, 155.0, 132.8, 91.3, 45.3, 37.5 ppm.
4.3 5-(1H-Pyrazol-3-yl)-pyrimidine (5)
A mixture of hydrazine hydrate (0.23 mL, 4.74 mmol) and acetic acid (0.28 mL, 4.84 mmol) in methanol (2 mL) was added drop wise to a solution of 4 (0.420 g, 2.37 mmol) in methanol (8 mL) at 0 °C. As after 5 h stirring at room temperature, traces of 4 were detected, an additional portion of reagents (10% of the initial amount) was added. After 7.5 h, the reaction mixture was diluted with CH2Cl2 (80 mL) and stirred with 5% aqueous Na2CO3 (20 mL). The aqueous layer was separated and extracted thoroughly with CH2Cl2 (40 × 20 mL). The crude product was purified by flash column chromatography (silica, 10% CH3OH in CH2Cl2) to afford 5 (0.320 g, 2.19 mmol) in 92% yield, mp 142-143 °C. Elemental analysis calcd (%) for C7H6N4 (146.15): C 57.53, H 4.14, N 38.34; found : C 57.63, H 4.15, N 37.76; 1H NMR (CDCl3, 500 MHz) δ 9.19 (s, 2 H, 4-H and 6-H), 9.08 (s, 1 H, 2-H), 7.79 (s, 1 H, 4′-H or 5′-H), 6.87 (d, 3JH,H = 2.4 Hz, 1 H, 5′-H or 4′-H), 3.31 (s, 1 H, NH) ppm; 13C NMR (CDCl3, 126 MHz) δ 157.1 (2-C), 153.9 (4-C and 6-C), 145.4, 130.8, 128.5, 103.1 ppm.
4.4 1,3,5-Tris [(pyrimidin-5′-yl)-3′-(1′H-pyrazol-1′-yl)methyl] benzene (1)
NaH (60% in oil, 0.055 g, 1.375 mmol) was added to a solution of pyrazole 5 (0.150 g, 1.026 mmol) in THF (5 mL) at 0 °C under dinitrogen. After 10 minutes stirring at room temperature a solution of 1,3,5-tribromomethylbenzene (0.117 g, 0.328 mmol) in THF (5 mL) was added to the reaction mixture, which was stirred for 24 h and subsequently quenched by addition of water (5 mL) at 0 °C. The slurry was diluted with water (30 mL) and extracted into CH2Cl2 (3 × 20 mL). The crude product was purified by column chromatography (standard alumina, 1% CH3OH in CH2Cl2) to afford tripod 1 (0.101 g, 0.0173 mmol) in 53% yield, based on tribromomethylbenzene, mp 196-197 °C. Elemental analysis calcd (%) for C30H24N12·CH3OH (584.25): C 63.80, H 4.84, N 28.80; found C 63.44, H 4.40, N 28.96; 1H NMR (CDCl3, 600 MHz, 298 K) δ 9.14 (s, 3 H, 2-H), 9.05 (s, 6 H, 4-H and 6-H), 7.46 (d, 3JH,H = 1.8 Hz, 3 H, 5′-H), 7.03 (s, 3 H, H′), 6.62 (d, 3JH,H = 1.8 Hz, 3 H, 4′-H), 5.34 (s, 6 H, CH2) ppm; 1H NMR (d6-dmso, 600 MHz, 298 K) δ 9.07 (s, 3 H, 2-H), 9.06 (s, 6 H, 4-H and 6-H), 7.94 (d, 3JH,H = 2.4 Hz, 3 H, 5′-H), 7.08 (s, 3 H, H′), 6.88 (d, 3JH,H = 2.4 Hz, 3 H, 4′-H), 5.40 (s, 6 H, CH2) ppm; 13C NMR (CDCl3, 151 MHz, 298 K) δ 157.7 (2-C), 153.7 (4-C and 6-C), 146.0 (3′-C), 138.1 (CH2C), 131.7 (5′-C), 127.4 (5-C), 126.3 (CH′), 104.0 (4′-C), 55.8 (CH2) ppm.
4.5 1,3,5-Tris [(pyrimidin-5′-yl)-3′-(1′H-pyrazol-1′-yl)methyl]-2,4,6-triethylbenzene (2)
Prepared as described above for 1 from NaH (60% in oil, 0.10 g, 2.50 mmol), pyrazole 5 (0.300 g, 2.053 mmol) and 1,3,5-tribromomethyl-2,4,6-triethylbenzene (0.290 g, 0.658 mmol). The reaction was quenched after 5 h stirring at room temperature. The crude product was purified by repeated flash column chromatography (silica 35-70 μm, 3-10% gradient CH3OH in CH2Cl2) to afford tripod 4 (0.335 g, 0.519 mmol) in 79% yield, based on tribromomethyltriethylbenzene, mp 147-148 °C. Elemental analysis calcd (%) for C36H36N12·0.5 H2O (645.77) : C 66.96, H 5.77, N 26.03; found C 66.82, H 5.75, N 25.71; 1H NMR (CDCl3, 600 MHz, 298 K) δ 9.13 (s, 3 H, 2-H), 9.10 (s, 6 H, 4-H and 6-H), 7.13 (d, 3JH,H = 2.1 Hz, 3 H, 5′-H), 6.60 (d, 3JH,H = 2.1 Hz, 3 H, 4′-H), 5.53 (s, 6 H, CH2), 2.81 (q, 3JH,H = 7.7 Hz, 6 H, CH2′), 1.07 (t, 3JH,H = 7.7 Hz, 9 H, CH3′) ppm; 1H NMR (d6-dmso, 600 MHz, 298 K) δ 9.14 (s, 6 H, 4-H and 6-H), 9.10 (s, 3 H, 2-H), 7.62 (d, 3JH,H = 2.4 Hz, 3 H, 5′-H), 6.92 (d, 3JH,H = 2.4 Hz, 3 H, 4′-H), 5.51 (s, 6 H, CH2), 2.86 (q, 3JH,H = 7.5 Hz, 6 H, CH2′), 0.92 (t, 3JH,H = 7.5 Hz, 9 H, CH3′) ppm; 13C NMR (CDCl3, 151 MHz, 298 K) δ 157.7 (2-C), 153.7 (4- and 6-C), 146.6 (CH2C and CH2′C), 145.8 (3′-C), 130.2 (5′-C), 127.5 (5-C), 103.5 (4′-C), 50.1 (CH2), 23.61 (CH2′), 15.59 (CH3′) ppm.
4.6 [Pd(1)(bipy)](NO3)2
A solution of [Pd(bipy)(ONO2)2] (0.0141 g, 3.65 × 10–5 mol) in d6-dmso was added to a suspension of ligand 1 (0.021 g, 3.80 × 10–5 mol) in d6-dmso, the final volume being 1 mL. The resulting clear yellow solution was evaporated to dryness under vacuum at 60 °C. The residue was dissolved in acetone/H2O. The solid material that precipitated upon further addition of acetone was collected, washed with acetone and dried at 35 °C under vacuum. Yield: 0.0291 g (74%). Elemental analysis calcd (%) for C40H32N16O6Pd·2H2O·C3H6O (1033.33) : C 49.98, H 4.10, N 21.69; found C 49.66, H 4.00, N 21.80; 1H NMR (d6-dmso, 600 MHz, 293 K) δ 10.06 (s, 2 H, 6-H), 9.78 (s, 2 H, 2-H), 9.40 (d, 4JH,H = 1.8 Hz, 2 H, 4-H), 9.16 (s, 2 H, 4°-H, 6°-H), 9.12 (s, 1 H, 2°-H), 8.79 (d, 3JH,H = 7.8 Hz, 2 H, δ-H), 8.52 (ddd, 3JH,H = 7.8 Hz, 3JH,H = 7.8 Hz, 4JH,H = 1.2 Hz, 2 H, γ-H), 8.14 (d, 4JH,H = 1.8 Hz, 2 H, 5′-H), 7.99 (s, 1 H, 5′′-H), 7.84 (d, 3JH,H = 5.7 Hz, 2 H, α-H), 7.71 (s, 1 H, H′′), 7.69 (ddd, 3JH,H = 7.8 Hz, 3JH,H = 5.7 Hz, 4JH,H = 1.2 Hz, 2 H, β-H), 7.47 (s, 2 H, H′), 7.02 (d, 4JH,H = 1.8 Hz, 2 H, 4′-H), 6.97 (s, 1 H, 4′′-H), 5.38 (s, 2 H, CH2°), 5.37 (AB, JAB = 14.1 Hz, ΔνAB = 115 Hz, 4 H, CH2) ppm; 13C NMR (d6-dmso, 151 MHz, 293 K) δ 157.3 (2-C, 2°-C), 156.6 (4-C), 155.7 (ɛ-C), 153.2 (4°-C, 6°-C), 152.8 (6-C), 151.0 (α-C), 144.9 (3′′-C), 143.3 (3′-C), 142.8 (γ-C), 138.0 (C°), 137.4 (C), 132.7 (5′-C, 5′′-C), 130.6 (C′′), 129.8 (5-C), 128.6 (β-C), 128.3 (C′), 127.0 (5°-C), 124.6 (δ-C), 104.5 (4′-C), 104.0 (4′′-C), 55.3 (CH2), 54.9 (CH2°) ppm.
4.7 [Pd(2)(bipy)](NO3)2
A solution of [Pd(bipy)(ONO2)2] (0.0122 g, 3.16 × 10–5 mol) in d6-dmso was added to a suspension of ligand 2 (0.0202 g, 3.16 × 10–5 mol) in d6-dmso, the final volume being 1 mL. The resulting clear yellow solution was evaporated to dryness under vacuum at 60 °C. The residue was triturated in acetone/H2O, and the solvents evaporated to dryness, leaving a pale yellow powder, which was further dried under vacuum at 50 °C. Yield: 0.0302 g (84%). Elemental analysis calcd (%) for C46H44N16O6Pd·2H2O·C2H6OS (1137.55) : C 50.68, H 4.78, N 19.70, S 2.82; found C 50.81, H 4.89, N 19.25, S 1.70; 1H NMR (d6-dmso, 600 MHz, 293 K) δ 9.96 (d, 4JH,H = 2.1 Hz, 2 H, 6-H), 9.84 (s, 2 H, 2-H), 9.39 (d, 4JH,H = 2.1 Hz, 2 H, 4-H), 9.08 (br s, 1 H, 2°-H), 9.03 (s, 2 H, 4°-H, 6°-H), 8.80 (dd, 3JH,H = 7.8 Hz, 4JH,H = 1.2 Hz, 2 H, δ-H), 8.51 (ddd, 3JH,H = 7.8 Hz, 3JH,H = 7.8 Hz, 4JH,H = 1.2 Hz, 2 H, γ-H), 8.36 (d, 4JH,H = 1.8 Hz, 2 H, 5′-H), 7.73 (d, 3JH,H = 2.4 Hz, 1 H, 5′′-H), 7.69 (ddd, 3JH,H = 7.8 Hz, 3JH,H = 5.7 Hz, 4JH,H = 1.2 Hz, 2 H, β-H), 7.55 (dd, 3JH,H = 5.7 Hz, 4JH,H = 1.2 Hz, 2 H, α-H), 7.04 (d, 3JH,H = 1.8 Hz, 2 H, 4′-H), 6.87 (d, 3JH,H = 1.2 Hz, 1 H, 4′′-H), 5.55 (AB, JAB = 15.0 Hz, ΔνAB = 45 Hz, 4 H, CH2), 5.36 (s, 2 H, CH2°), 4.18 (q, 3JH,H = 7.8 Hz, 2 H, CH2′′), 2.98 (m, 4 H, CH2′), 1.33 (t, 3JH,H = 7.8 Hz, 3 H, CH3′′), 0.78 (t, 3JH,H = 7.2 Hz, 3 H, CH3′) ppm; 13C NMR (d6-dmso, 151 MHz, 293 K) δ 157.4 (2-C), 157.2 (2°-C), 156.9 (4-C), 155.6 (ɛ-C), 153.5 (6-C), 153.0 (4°-C, 6°-C), 150.5 (α-C), 147.0 (C′′), 144.9 (C′), 144.3 (3′′-C), 143.1 (3′-C), 142.9 (γ-C), 134.0 (5′-C), 132.1 (5′′-C), 131.3 (C), 130.3 (5-C), 129.9 (C°), 128.8 (β-C), 127.0 (5°-C), 124.7 (δ-C), 103.8 (4′-C), 103.5 (4′′-C), 49.0 (CH2, CH2°), 23.9 (CH2′′), 22.2 (CH2′), 15.8 (CH3′′), 15.7 (CH3′) ppm.
5 Conflicts of interest
There is no conflict of interest affecting the corresponding authors and his co-authors.
Acknowledgments
The French Ministry of Higher Education and Research is thanked for a postdoctoral fellowship to PPM. Dr. Michel Meyer is gratefully acknowledged for helpful discussions.
Appendix B Supplementary data
Structure of the initial target complexes ([Pd3(bipy)3(1)]6+ and [Pd3(bipy)3(2)]6+). Proton and carbon labeling of 1, 2 and their [Pd(bipy)]2+ complexes. Table of 1H NMR data of [Pd(bipy)](NO3)2, 1, 2, and their [Pd(bipy)]2+ complexes, VT 1H NMR spectra of [Pd(1)(bipy)](NO3)2 and [Pd(1)(bipy)](NO3)2, 1H/1H NMR COSY and ROESY of [Pd(1)(bipy)](NO3)2, 13C/1H NMR HSQC and HMBC of [Pd(1)(bipy)](NO3)2. Table of estimated errors for ΔG‡’s.
1 DMSO turned out to be the only solvent in which homogeneous solutions could be obtained after mixing the reactants. Neither water, nor MeOH and acetone did lead to comparable results. However, [Pd(1)(bipy)](NO3)2 is soluble in acetone/water mixtures, whereas [Pd(2)(bipy)](NO3)2 is not.