

1 Introduction
Steroid hormones are of extraordinary physiological importance. Sexual hormones (androgens, estrogens) regulate the development of secondary sexual characteristics, glucocorticoids such as cortisol are involved in the stress response of mammals and mineralocorticoids such as aldosterone regulate the blood pressure due to their role in modulating the water and salt housekeeping of the body [1–6]. Generally, their effects are mediated through specific nuclear receptors. Besides this classic way of affecting the cellular gene expression, steroid hormones also exert part of their action via non-genomic mechanisms [7–11].
In the biosynthesis of steroid hormones, several cytochromes P450 are involved. Since many pathological conditions are associated with increased or decreased levels of specific steroid hormones, these enzymes are promising therapeutic targets, and their selective regulation is highly desirable in order to influence the amounts of steroid hormones produced [12–17].
Aldosterone, the most important mineralocorticoid, has been associated with hypertension, fibrosis of the heart, and severe heart failure [18–21]. Although the mechanisms of these damaging effects are, as yet, poorly understood they may at least partly be mediated through non-genomic pathways [22–26]. To focus on the investigation of these non-genomic actions of aldosterone, a model system has been developed in our group to study the effects of aldosterone and other steroid hormones without interference of steroid hormone nuclear receptors. We chose the fission yeast Schizosaccharomyces pombe (S. pombe), which does not contain any nuclear steroid receptors, but many genes and regulatory mechanisms that are close to those of mammals [27,28].
The differential protein patterns in S. pombe upon mineralocorticoid administration were studied using 2D-gel electrophoresis and mass spectrometry. Eleven proteins that were differentially regulated by aldosterone [29] and 19 proteins that were specifically affected by the aldosterone-precursor 11-deoxycorticosterone (DOC) [30] have been identified. Four of these proteins showed altered levels in both cases [31] and therefore may be specifically associated with non-genomic actions, which makes them potential targets for the development of new drugs (Table 1).
Differentially regulated proteins by both aldosterone (Aldo) and by DOC. The average intensities (absolute and relative values) of the spots containing the indicated proteins on the respective 2D-gels are listed. NAD-dependent malic enzyme and glyceraldehyde-3-phosphate dehydrogenase 1 were present in two different spots each. The spot intensity data are taken from Böhmer et al. (2006) [29] (Aldo) and estimated according to Hwang (2007) [30] (DOC), respectively. Upregulation (increased intensity) is indicated by ↑ arrows, downregulation (decreased intensity) is indicated by ↓ arrows. n.d., not detectable.
| Protein name | S. pombe gene | Average spot intensities | Regulation by | ||||||||
| DOC control | DOC sample | Aldosterone control | Aldosterone sample | DOC | Aldo | ||||||
| Absolute [ppm] | Relative (%) | Absolute [ppm] | Relative (%) | Absolute [ppm] | Relative (%) | Absolute [ppm] | Relative (%) | ||||
| Enolase 1-1 | eno101 | 26,000 | 100% | 53,000 | 203.9% | 1102.9 | 100% | 314.8 | 28.5% | ↑ | ↓ |
| NAD-dependent malic enzyme | mae2 | 42,000 | 100% | 7,000 | 16.7% | 2666.0 | 100% | 1114.7 | 41.8% | ↓ | ↓ |
| 23,000 | 100% | 6,000 | 26.1% | ||||||||
| Glyceraldehyde-3-phosphate dehydrogenase 1 | tdh1 | 88,000 | 100% | 13,000 | 14.8% | 207.4 | 100% | n.d. | n.d. | ↓ | ↓ |
| 48,000 | 100% | 22,500 | 46.9% | ||||||||
| Protein vip1 | vip1 | 49,000 | 100% | 23,000 | 46.9% | 1796.4 | 100% | 259.5 | 14.5% | ↓ | ↓ |
Among the latter, the protein vip1 seems to be of special interest. This protein first had been identified using a polyclonal p53-antiserum while searching for a potential homologue of the human tumour suppressor protein p53 in S. pombe [32], which does not possess any known p53-homologues itself [28]. p53 is of crucial importance for the conservation of the genomic integrity in higher eukaryotes [33–36]. Loss of p53-activity in a cell is frequently the first step in the development of tumours that may become a threat to the existence of the entire organism.
In the C-terminal area covering 34 amino acids, vip1 displays a 50% sequence homology to a part of the so-called prolin-rich domain of human p53 [32] (Fig. 1A). Further examinations in order to characterize the vip1 protein, however, showed that vip1 is most probably not a p53-homologue, but rather seems to be involved in the organization of the cytoskeleton [37]. Bioinformatic analysis of the vip1 nucleotide sequence revealed three potential domains in its protein structure: an N-terminal RNA-recognition motif (RRM) [38], a central area, and a C-terminal region containing two PEST-motifs as potential binding sites for ubiquitin [37] (Fig. 1A). Besides the similarities in the central domain between vip1 and myosin, it could be shown that tubulin and vip1 are capable of binding to each other in vitro. In addition, co-localization of green fluorescence protein (GFP)-labelled vip1 to the microtubules during cell division in vivo could be demonstrated. On the basis of findings in cold-shock experiments using GFP-vip1, the possible participation of this protein in reconstitution of a functional microtubule framework was corroborated [37]. However, little is known about the functionality of this 27.5 kDa protein so far.
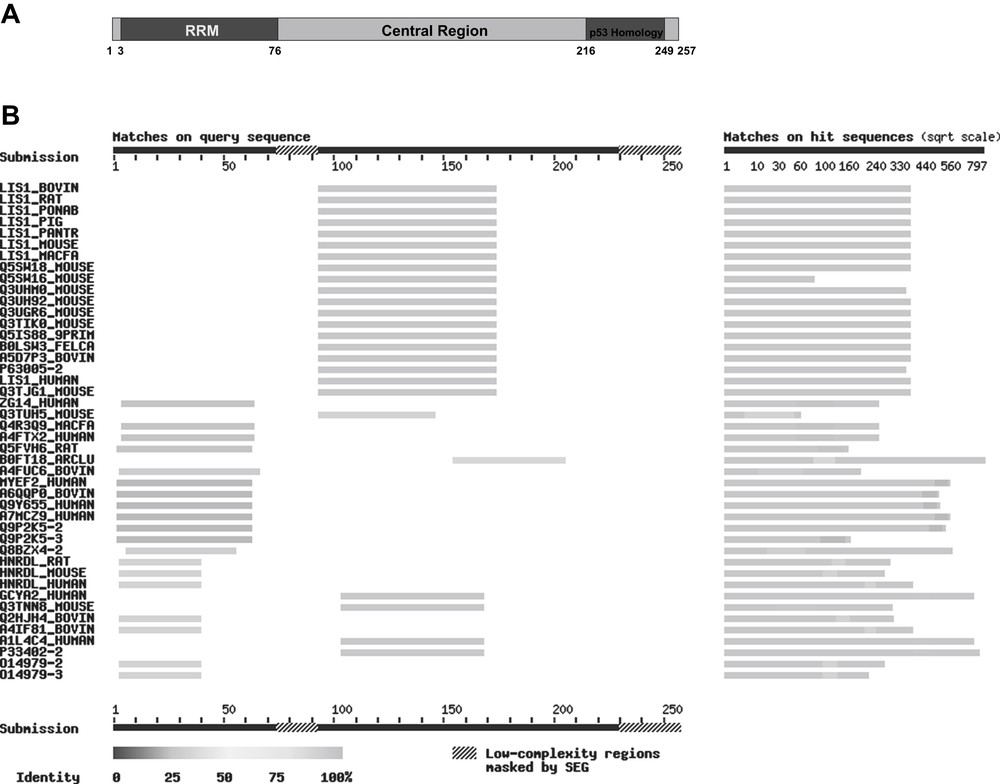
(A) Putative domain structure of the S. pombe protein vip1 and homologies with other proteins: N-terminal: RNA recognition motif (RRM, dark grey), amino acids 3–76, homologies with RNA-binding proteins. Central region (grey), amino acids 77–215: homologies with mammalian LIS1 proteins. C-terminal (patterned): homology with the human p53 tumour suppressor protein (amino acids 216–249). The numbers indicate the amino acid positions within the vip1 sequence. (B) BLAST search for homologues of vip1 in mammals. The picture was taken from SIB BLAST Network Service (ExPASy) (www.expasy.org). In this application, the query sequence is run through the program SEG for filtering low-complexity regions (“masked by SEG”) (e.g. stretches of cysteine, hydrophobic regions in membrane proteins), which tend to produce spurious, insignificant matches with sequences in the database that have the same kind of low-complexity regions, but are unrelated biologically.
As aldosterone is known to induce fibrosis in various tissues [18,19,39] and since fibrosis is always associated with an increase in connective tissue [40,41], it is interesting that aldosterone has been shown to reduce the amount of vip1 [29,31], which seems to be associated with the cytoskeleton and thus with scleroproteins that are known to be involved in the function of connective tissues. The mechanism underlying the aldosterone-mediated action of vip1 are completely unknown at present. To get a deeper insight into this function, expression patterns and interacting partners of vip1 have to be identified.
The work described here therefore aimed at characterizing the S. pombe protein vip1 and expressing it heterologously in the bacterial host Escherichia coli (E. coli) in order to obtain indications on its function and sufficient amounts of the protein for further investigations.
2 Results and discussion
2.1 Bioinformatic analysis
To get more information about possible functions of vip1 in S. pombe, we first performed a bioinformatic analysis. As information about protein sequences has been rapidly growing since the last investigations concerning vip1 in 2000 [32,37], we intended to confirm their results mentioned in the introduction and to obtain additional clues about the possible function of vip1 as well as its molecular structure. We were looking for potential homologues of vip1 and for related proteins, particularly in mammals. The SIB BLAST Network Service (ExPASy) (server developed at SIB, NCBI BLAST 2 software) (www.expasy.org) was used to search the Swiss-Prot database for this purpose.
Homologies of actual or hypothetical RNA-binding proteins to the N-terminal area of vip1, which contains the RNA-recognition motif (RRM), have been identified. It can be presumed that the similarities are due to a common RNA-binding function. Among these proteins are, for instance, the human gastric cancer antigen Zg14, the human and rat myelin expression factor 2 (MyEF-2), and the heterogeneous nuclear ribonucleoprotein d-like proteins (hnRPD-like proteins) of human, rat and mouse (Fig. 1B).
The central region of vip1 shows homologies with the human guanylate cyclase soluble subunit alpha-2 (GCS-alpha-2) and a similar mouse protein, but above all with several mammalian LIS1 (Lissencephaly-1) proteins, namely bovine, rat, mouse, pig, cynomolgus monkey, Sumatran orang-utan, chimpanzee, and also human LIS1 (Fig. 1B). This protein is formally named platelet-activating factor acetylhydrolase IB subunit alpha (PAF-AH alpha). The respective gene is deleted in patients with Miller-Dieker syndrome who develop a brain malformation called lissencephaly [42]. Interestingly, LIS1 interacts with tubulin and microtubules [43], is associated with the dynein/dynactin pathway and mediates the nucleus to the centrosome in neuronal migration [44,45].
To confirm the homology between vip1 and the human Lissencephaly-1 protein, we aligned the amino acid sequences of both proteins using EMBOSS Needle (http://www.ebi.ac.uk/Tools/emboss/align/index.html) [46]. The sequences showed 6.9% overall identity and 11.3% overall similarity (score: 62) (Fig. 2). As can be seen in Figure 2, the identity is, however, much higher when taking into account only amino acids 98 to 257 of vip1 and 1 to 141 of LIS1, where the sequences overlap. In this case, the proteins display 21.0% identity (37/176 residues) and even 34.1% similarity (60/176 residues) (Fig. 2).

Sequence alignment of the fission yeast protein vip1 (VIPI_SCHPO) and human LIS1 (LIS1_HUMAN) using the EMBOSS Needle tool. Vertical dashes: amino acid identities, dots: amino acid similarities. Overall identitiy: 6.9%, Similarity: 11.3%, Score: 62.
A homology model of the three-dimensional structure of vip1 has been obtained using the SWISS-MODEL Server of the ExPASy protein database (http://swissmodel.expasy.org) [47–49] and ModBase of the UCSF (University of California, San Francisco). The structure of the N-terminal area (residues 2 to 72 for SWISS-MODEL and 2 to 89 for ModBase) is typical of an α/β protein. It shows several putative α-helices and β-sheet structures. The template structure used by ModBase was the NMR structure of the N-terminal RRM domain of the cleavage stimulation factor 64 kDa subunit (RCSB PDB code 1p1tA) [50]. Other possible models, according to ModBase, use UP1, the two-RNA-recognition motif domain of hnRNP A1 (RCSB PCT code 1/3kA) [51] (residues 1 to 71 of vip1) and the cap binding complex (RCSB PCT code 1n52B) [52] (residues 2 to 101 of vip1) as template proteins (data not shown). For the central and C-terminal parts of vip1 no structure was derived from these servers.
The existence of the N-terminal RNA-recognition motif (RRM) could thus be confirmed. As for the central region of vip1, we discovered a homology with the human Lissencephaly-1 protein, which interacts with tubulin and microtubules. This finding, together with the similarities in the central domain between vip1 and myosin and its ability to bind tubulin in vitro [37], corroborates the assumption that vip1 might be involved in the organization of the cytoskeleton. A homology model of the three-dimensional structure of the N-terminal part of vip1 showing several putative α-helices and β-sheet structures could been obtained, whereas the structures of the central and C-terminal areas of vip1 remain unclear.
2.2 Cloning, expression and purification of vip1
Since the vip1 gene (774 bp) does not contain any introns, the DNA could be amplified directly from the S. pombe genome through PCR using gene-specific primers. In order to simplify the protein purification procedure after the expression, a His6-tag (consisting of 6 histidine residues) has been attached to the vip1 protein. We used the restriction endonucleases SacII and KpnI to clone the vip1-cDNA into the multiple cloning site of the expression vector pASK-IBA37plus, which links the N-terminus of the insert via a factor Xa cleavage site to the nucleotide sequence coding for the 6xHistidine-tag (Fig. 3). The restriction sites of both enzymes do not occur in the original vip1 sequence and are found only once in the vector DNA, namely within the multiple cloning site. The expression cassette of the plasmid is under transcriptional control of a tetracycline promoter/operator. In addition, the vector contains an ampicillin resistance gene.
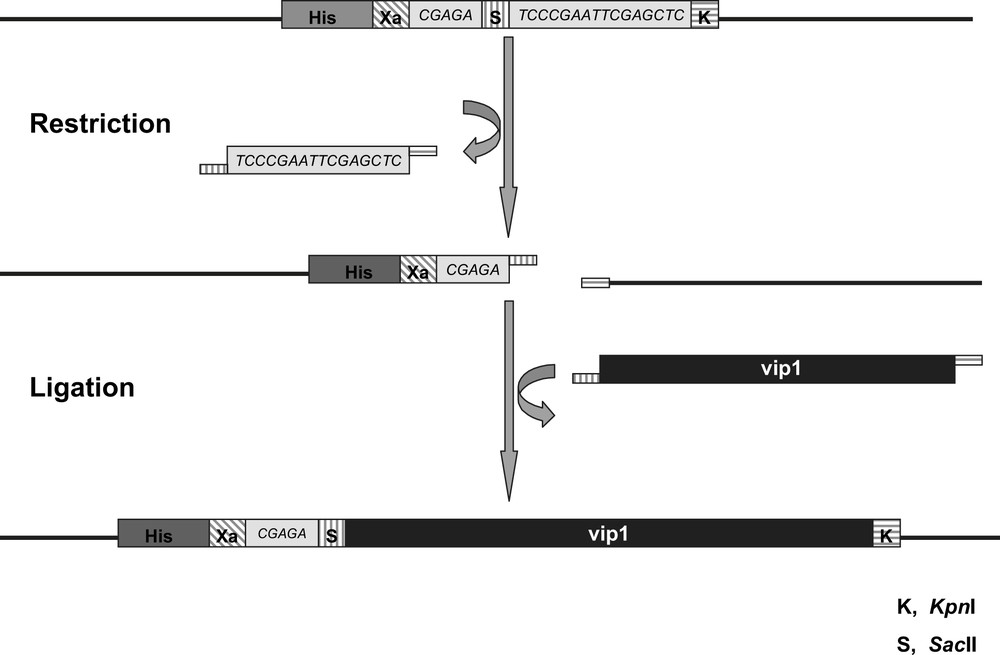
Schematic view of the Multiple Cloning Site (MCS) of the vector pASK-IBA37plus. The vip1-cDNA (black bar) is inserted by cleaving the SacII (S, vertically striped) and KpnI (K, horizontally striped) restriction sites, respectively, followed by ligation of the vector and the insert cDNAs. The sequences coding the 6xHistidine-tag (His, dark grey) and the factor Xa cleavage site (Xa, diagonally striped) are part of the vector backbone 5′ of the vip1-cDNA insert, from which they are separated by a short linker sequence (light grey). The lengths of the different sequences are not to scale.
After ligation of the vector backbone with the vip1-cDNA, the plasmid was introduced into E coli cells by transformation of the bacteria. In the logarithmic growth phase of the bacteria, the vector's tetracycline promoter was activated by adding anhydrotetracycline hydrochloride (AHT) to the medium of the main culture, which resulted in the heterologous over-production of the His-tagged vip1 protein. The effect on the protein pattern of the host organism, E. coli BL21, is depicted in Fig. 4A. Analysis of the protein expression by sodium dodecyl phosphate polyacrylamide gel electrophoresis (SDS-PAGE) clearly showed a protein band with a size of approximately 30 kDa after the expression as compared with the cultures before induction (Fig. 4A). In order to separate the protein of interest from the other E. coli proteins, i.e. to purify it, the cell lysate has been subjected to Immobilized Metal Affinity Chromatography (IMAC), taking advantage of the specific binding of the 6xhistidine affinity tag to Co2+ ions. Fig. 4B shows how most of the non-vip1-bands in the gel, and thus the corresponding proteins, have disappeared as opposed to Fig. 4A. However, there are still some bands present in the lower molecular-weight range which do not represent the native vip1 protein. In order to remove the remaining undesired proteins, Ion Exchange Chromatography (IEC) has been performed as a second purification step. Finally, vip1 was available as an almost pure protein (Fig. 4C).
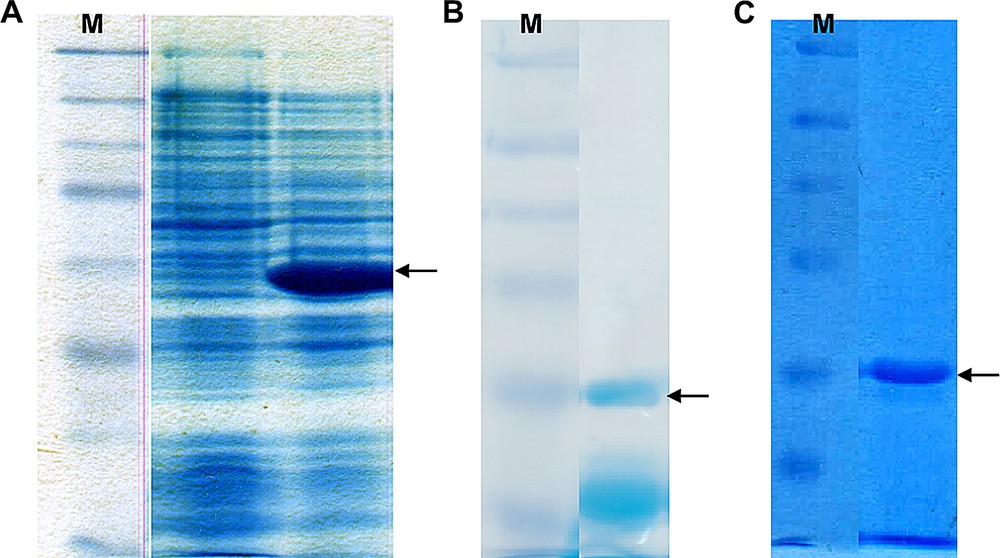
(A) 12.5% SDS polyacrylamide gel showing the E. coli cell lysates before induction of the heterologous vip1 expression (left) and after the vip1 expression (right). The arrow indicates the 30-kDa putative vip1 band, which is clearly visible after protein expression. M, protein standard (marker). (B) 10% SDS gel showing a typical elution fraction after IMAC purification of the cell lysate. Besides the vip1 band indicated by the arrow, there are still some other bands in the lower molecular weight (MW)-range, which may represent contaminations or degradation products. M, protein standard. (C) 10% SDS gel showing a representative elution fraction. As a result of the IEC purification step, the lower MW-bands could be removed. The arrow indicates vip1. M, protein standard.
2.3 Mass spectrometry analysis
The identity of the purified protein could be confirmed using MALDI-TOF/TOF mass spectrometry (Fig. 5A) and tandem mass spectrometry (Fig. 5B). The theoretical peptide masses of vip1, their sequences and their positions within the protein according to PeptideMass (ExPASy) are listed in Table 2. All the theoretical peptides (with a mass of >500 Da) together cover 96.5% of the vip1 sequence. The theoretical isoelectric point (pI) of vip1 is 5.54, its theoretical molecular weight amounts to 27,459.24 (average mass) and 27,442.82 (monoisotopic mass). There are five putative phosphorylation sites within the vip1 sequence (positions 132, 177, 230, 232, and 235). (Data taken from PeptideMass (ExPASy)).
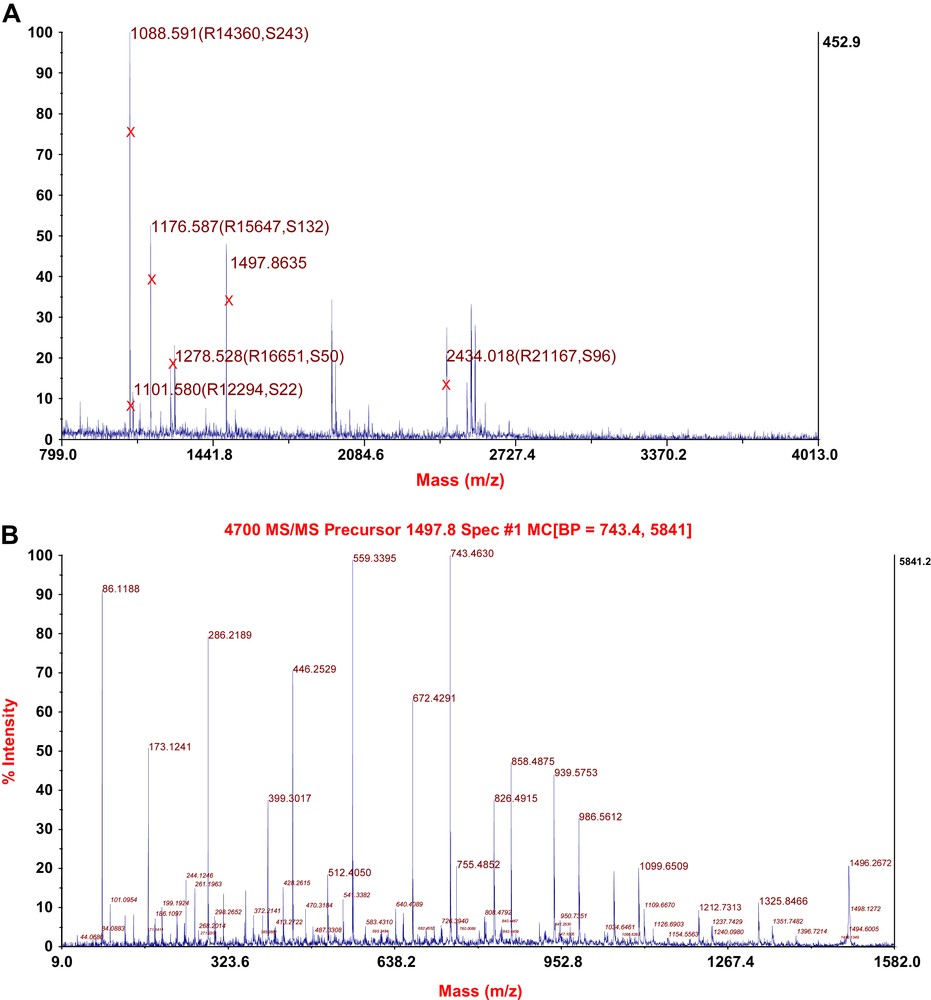
(A) MALDI-TOF spectrum of the protein vip1. Masses marked by X represent peptide fragments of vip1. R, Resolution. S, Signal to noise-ratio. (B) Tandem mass spectrometry (MS/MS) spectrum of the vip1-specific peptide fragment with the mass of 1497.8635 (see (A) and Table 2).
Theoretical peptide masses, positions, and sequences after tryptic digestion of vip1 according to PeptideMass (ExPASy). Those peptides which could be identified in the MALDI-TOF experiment are indicated in the rightmost column.
| Mass | Position | Peptide sequence | Identified by MS |
| 2434.1076 | 69–94 | IQITSEDGGAASTTDQGGAGGDQAAR | yes |
| 1914.9039 | 225–244 | NQAEGTASPASSTPTAPAEK | – |
| 1888.9684 | 1–17 | MSNQVIVTNISPEVTEK | – |
| 1497.8635 | 55–68 | TALLLQDALLGQNK | yes |
| 1411.7063 | 122–134 | SIQLDQSYGVSSK | yes |
| 1278.5823 | 18–28 | QISDFFSFCGK | yes |
| 1269.6474 | 175–185 | TSSLVSTYFHK | yes |
| 1264.6756 | 208–218 | NQLLGIHNEAR | – |
| 1261.6423 | 111–121 | GYHLSDVTLEK | yes |
| 1204.6168 | 186–198 | ALETAAGTSAGQK | – |
| 1176.6371 | 45–54 | IQFERPSATK | yes |
| 1101.6262 | 137–147 | GILESALSGVR | yes |
| 1088.6310 | 101–110 | SAIISELLSR | yes |
| 1060.5157 | 245–254 | EPTAPTTESK | – |
| 1023.4741 | 199–207 | VQNAYEDGK | – |
| 877.4625 | 29–36 | VSNISTEK | yes |
| 821.3999 | 37–44 | SGETQTAK | yes |
| 789.3737 | 159–165 | ANEVDNK | – |
| 776.3937 | 153–158 | YHVTEK | – |
| 772.3948 | 95–100 | QEDKPR | – |
| 680.3613 | 166–171 | FAISDK | – |
| 604.3049 | 148–152 | SVNER | – |
| 517.2980 | 220–224 | LADAK | – |
The experimental data show a sequence coverage of 51% by peptide mass fingerprinting (PMF) and 28% by tandem mass spectrometry (MS/MS) (Fig. 6). It is evident that the total coverage can is then 51% since all MS/MS spectra rely on peptides found by PMF (Fig. 6). The theoretical peptides that could be identified experimentally by either method are indicated in Table 2.

vip1 sequence coverage of the mass spectrometric analysis. Underlined boldface letters in italics: fragments detected by peptide mass fingerprint (PMF). Peptides detected by tandem mass spectrometry (MS/MS) are shaded in dark grey. Neighbouring peptides are separated by slashes.
Taken together, we were able to heterologously express vip1 from S pombe in E coli. The protein was purified from there taking advantage of the introduced His-Tag using an affinity chromatography followed by Ion Exchange Chromatography. The identity of the purified protein was confirmed by mass spectrometry. Bioinformatic analyses indicated that vip1 is homologous to the human LIS1, which was shown to interact with proteins from the cytoskeleton. Further analyses using antibodies produced against vip1 are now under way to study the expression of this protein in dependence on aldosterone and other steroid hormones and to find possible interaction partners of this protein.
3 Materials and methods
3.1 Materials
All chemicals used were of the highest quality available, i.e. analytical grade or higher quality. Nutrient broth and nutrient broth agar were obtained from SIFIN (Institut für Immunpräparate und Nährmedien GmbH, Berlin, Germany), lysogeny broth (LB) medium was from BD (Becton, Dickinson and Company, Sparks, Maryland, U.S.A.). Yeast extract (YE) medium was prepared as described by Moreno et al. (1991) [53]. Components for yeast culture media were purchased from BD, Acros Organics (Geel, Belgium), and from Roth (Carl Roth GmbH & Co. KG, Karlsruhe, Germany). Dithioerythritol (DTE), ammonium bicarbonate and sorbitol were from Sigma (Sigma–Aldrich, Inc., St. Louis, Missouri, U.S.A). Ethylenediaminetetraacetic acid (EDTA), Tris, ethanol (HPLC grade), ampicillin (sodium salt) and Roti-Phenol/C/I were obtained from Roth. 3-methyl-1-butanol, sodium dihydrogen phosphate (anhydrous) and sodium phosphate dibasic dihydrate were provided by Fluka (Buchs, Switzerland). Phenylmethylsulfonylfluoride (PMSF) and sodium dodecyl sulphate (SDS) were supplied by Serva (Serva Electrophoresis GmbH, Heidelberg, Germany). Sodium citrate, Tris-HCl, potassium acetate, sodium acetate and imidazole (for synthesis) were obtained from Merck (Darmstadt, Germany).
Oligonucleotides were procured by MWG Biotech AG (now Eurofins MWG Operon, Ebersberg, Germany). Pfu DNA polymerase, the restriction endonucleases SacII and KpnI, as well as trypsin were purchased from Promega (Madison, Wisconsin, U.S.A.).
3.2 Preparation of genomic DNA from Schizosaccharomyces pombe
S. pombe wild-type strain h−S L 972 was kindly provided by N. Kaeufer, TU Braunschweig, Germany. The cells were cultured on yeast extract agar (YEA) plates at 30 °C. For the preparation of the genomic DNA, S pombe was grown in yeast extract (YE) medium at 30 °C (10 ml pre-culture in a 50 ml reaction tube for 24 h while shaking at 170 rpm, 100 ml main culture in a 250 ml Erlenmeyer flask inoculated with 10 ml of pre-culture at 170 rpm until a cell density of 107 cells/ml is reached). The cells were harvested by centrifugation of the main culture at 5000g for 5 min and then resuspended in 10 ml CIPS buffer (1.2 M sorbitol, 50 mM sodium citrate, 50 mM sodium phosphate (pH 5.6), 40 mM ethylenediaminetetraacetic acid (EDTA) in H2O).
The preparation of genomic DNA was carried out as described by Andreas Jungbluth [37]. The yeast cells were disrupted by addition of 2.5 mg zymolyase and incubation at 37 °C for 30 to 60 min (until the majority of the cells became spheroblasts). After centrifugation at 800g for 5 min, the pellet was resuspended in 4.5 ml 5x TE (Tris/EDTA) buffer (50 mM Tris–HCl (pH 7.5), 5 mM EDTA in H2O, autoclaved), and 500 μl of 10%-SDS (sodium dodecyl sulphate) solution were added. After incubation for 5 min at 65 °C the SDS was removed (together with the denatured and precipitated proteins) by addition of 1.5 ml of 5 M potassium acetate, incubation on ice for 30 min, and centrifugation two times for 10 min at 30,000 rpm in the HITACHI ultracentrifuge. The supernatant was mixed with an equal volume of isopropanol and incubated on ice for 5 min, then centrifuged at 5400g for 15 min in order to collect the precipitated nucleic acids. The pellet was washed with 1 ml of 70%-ethanol and dissolved in 2.5 ml 5x TE buffer. The RNA was removed by addition of 200 μg RNase A and incubation at 37 °C for one hour. The RNase and remaining proteins were then extracted and separated from the genomic DNA. The extraction was carried out with 2.5 ml of a phenol/chloroform/3-methyl-1-butanol mixture (25:24:1) (Roti-Phenol/C/I, Roth), then with 2.5 ml chloroform/3-methyl-1-butanol (24:1), and at last with 2.5 ml chloroform. In each step, the mixture was vortexed for 1 min and then centrifuged at 5400g for 5 min to separate the phases. The aqueous (upper) phase was then transferred to a new reaction tube and passed on to the next extraction step. After the last step, 250 μl 3 M sodium acetate (pH 5.5) and 7.5 ml pure ethanol were added to the aqueous phase, which was after that incubated at −20 °C for at least one hour in order to precipitate the genomic DNA. The DNA was then collected by centrifugation at 5400g for 30 min, washed with 200 μl 70%-ethanol and air-dried for 5 min. The purified genomic DNA was finally dissolved in 300 μl TE buffer and stored at −20 °C.
3.3 Bacterial strains, plasmid and DNA techniques
E coli strain TOP10F' (Invitrogen, Carlsbad, California, U.S.A.) (F'[lacIq Tn10(tetR)] mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacX74 deoR nupG recA1 araD139 Δ(ara–leu)7697 galU galK rpsL(StrR) endA1 λ−) was used for plasmid propagation. E. coli BL21 (B F−, ompT, hsdS (rB−, mB−), gal, dcm) cells and vector pASK-IBA37plus (IBA GmbH, Göttingen, Germany) were utilised for heterologous protein expression.
The following primers were used to amplify the vip1 gene from the isolated S. pombe genomic DNA via a polymerase chain reaction (PCR) using Pfu DNA polymerase (Promega): 5′- GTT GAA ACC CGC GGA ATG AGC AAC CAA GTT ATT G –3′ (forward, at the 5′ end) and 5′- CAA GGT ACG GTA CCT TAC TCA GTG GTC TTT GAC -3′ (reverse, at the 3′ end). Underlined letters represent the SacII and KpnI endonuclease restriction sites, which were introduced upstream of the vip1 start codon and downstream of the vip1 stop codon, respectively.
The expression plasmid pASK-IBA37plus was also used as the vector backbone for the cloning of the vip1-cDNA. The expression cassette of the vector is under transcriptional control of the tetracycline promoter/operator. The plasmid confers ampicillin resistance to the host organism. Upon expression, a 6xHistidine-tag and a factor Xa cleavage site are fused to the N-terminus of the recombinant protein.
In order to clone the vip1-cDNA into the multiple cloning site of pASK-IBA37plus, both the vector and the PCR product obtained were digested with SacII and KpnI endonucleases. The compatible ends of the linearised vector backbone and the PCR product were then ligated with each other using the Fast-Link DNA Ligation Kit (Epicentre Biotechnologies, Madison, Wisconsin, U.S.A.) according to the manufacturer's instructions.
E. coli TOP10F' cells were transformed with the ligation product by means of electroporation at 1800 V using an Equibio EasyjecT Prima electroporator (Wolf Laboratories Ltd., Pocklington, York, U.K.) and plated on selective Nutrient Broth Agar I (NA I) plates containing 100 μg/ml ampicillin (final concentration). In order to select those cells which contain the correct plasmid construct, several of the colonies were screened by checking the characteristic DNA fragment sizes after isolating and cutting of the plasmids they contained. The plasmids were isolated by phenol/chloroform extraction followed by ethanol precipitation carrying out plasmid mini preparation using the NucleoBond PC 100 plasmid purification kit (Macherey-Nagel GmbH & Co. KG, Düren, Germany). The purified plasmids were then cut by SacII and KpnI, and the fragments were separated through DNA electrophoresis on a 1% agarose gel to check their lengths, i.e. to verify the presence of the insert. From the positive clones (the plasmids of which show the correct fragment sizes of vector and insert), the plasmids were amplified by growing the cells in 100 ml Nutrient Broth I (NB I) medium overnight and subsequent isolation of the plasmids by phenol/chloroform extraction and ethanol precipitation performing plasmid midi preparation using the NucleoBond PC 100 plasmid purification kit (see above). The sequence of the cloned gene was verified by DNA sequencing.
3.4 Protein expression and purification
Competent E. coli BL21 cells (50 μl) were transformed by means of the heat-shock method (42 °C for 45 s in a water bath) after adding 100 ng of the purified plasmid. The cells were suspended in 500 μl SOC (super optimal broth, catabolite repression) medium and incubated at 37 °C for 60 min on a desktop shaker for recovery. Four overnight cultures were prepared by inoculating 10 ml of sterile Lysogeny Broth (LB) medium containing 100 μg/ml ampicillin with 100 μl of the transformed cells for each culture and grown in 50 ml reaction tubes at 37 °C with shaking at 180 rpm overnight. Another 100 μl of the transformed cells were spread on a selective agar plate (100 μg/ml ampicillin) as a control. The next day, four main cultures containing 1 l sterile terrific broth (TB) medium with 100 μg/ml in a 2 l Erlenmeyer ridges flask each were inoculated by adding the overnight culture (10 ml) to each flask. The cultures were grown at 37 °C while shaking at 180 rpm.
Gene expression was induced by adding 200 μg/l (final concentration) anhydrotetracycline hydrochloride (AHT) (IBA GmbH, Göttingen, Germany) to the medium in each culture at approximately OD600 = 0.7. After the induction, additional ampicillin was added to a final concentration of 50 μg/ml in each flask, and the temperature of the culture was reduced to 28 °C. 16 h after induction the cells were harvested through centrifugation in a Beckman Coulter centrifuge at 4500 rpm for 10 min. The pooled pellets were stored in a 50 ml reaction tube at −20 °C.
Culture samples before induction and after expression were examined by sodium dodecyl phosphate polyacrylamide gel electrophoresis (SDS-PAGE).
The cell pellet was resuspended in buffer A (50 mM sodium phosphate buffer (pH 7.0) containing 300 mM sodium chloride and 10 mM imidazole) supplemented with 1 mM phenylmethylsulfonylfluoride (PMSF) (final concentration). Cells were disrupted by sonication in ice water using an Emich USD 30 sonicator (Emich Ultraschall GmbH, Berlin, Germany) (amplitude 25 Hz, bursts and cooling intervals for 30 s each (ratio 1:1), 20 min in total, four times), followed by centrifugation at 35,000 rpm, 4 °C for 30 min.
The vip1 protein was purified by immobilised metal affinity chromatography (IMAC) taking advantage of the N-terminally attached His6-tag. The cell lysate was loaded on a TALON Co2+ column (Clontech Laboratories, Inc., Mountain View, California, U.S.A.) previously equilibrated with buffer B (50 mM sodium phosphate buffer (pH 7.0) containing 300 mM sodium chloride). The column was washed with buffer B (alternatively with buffer A). Elution was performed using buffer C (buffer B containing 150 mM imidazole). Elution fractions were collected and their protein contents checked by SDS-PAGE. The fractions that showed the vip1-band were pooled and concentrated to a final volume of less than 10 ml using Amicon Ultra-15 centrifugal filter devices (Millipore, San Francisco, California, U.S.A.) with a nominal molecular weight limit of 10 kDa at 2000xg. The concentrated protein solution was diluted with potassium phosphate buffer (20 mM, pH 7.4) and concentrated again until most of the imidazole was removed from the solution (final volume less than 10 ml).
In order to remove the remaining undesired proteins, Ion Exchange Chromatography (IEC) was performed as a second purification step. The concentrated protein solution was loaded on a diethylaminoethyl cellulose (DEAE) anion exchange column (TOYOPEARL DEAE-650S, TOSOH Corporation, Tokyo, Japan) previously equilibrated with wash buffer (20 mM Tris, pH 7.5). The column was washed with the wash buffer, and afterwards the protein was eluted with elution buffer (20 mM Tris, pH 7.5, containing 500 mM NaCl), applying a linear gradient. Elution fractions were collected and their protein contents checked by SDS-PAGE. The fractions that showed the vip1-band were pooled and concentrated to a final volume of less than 10 ml while changing the buffer to potassium phosphate in order to remove the sodium chloride.
3.5 MALDI-TOF MS experiment
For the sample preparation, 2 mg of purified vip1 protein in potassium phosphate buffer were added to a fivefold volume of acetone and frozen overnight at −20 °C (acetone precipitation). The precipitated protein was spin down at 4 °C and 6000g for 5 min. The pellet was air-dried and re-dissolved in 1 ml urea buffer (50 mM Tris buffer (pH 8.0) containing 8 M urea and 4 mM dithioerythritol (DTE) (alternatively, dithiothreitol, DTT) in ultra-pure water (Milli-Q, Millipore, San Francisco, California, U.S.A.)), resulting in a protein concentration of approximately 2 mg/ml. The solution was incubated at 60 °C for one hour (denaturation) and then diluted 1:10 in ammonium bicarbonate solution (50 mM NH4HCO3 (pH 7.8) in ultra-pure water (Milli-Q)). Trypsin (500 ng/μl stock solution) was added to the protein solution in a ratio of 1:20. For the digestion to occur, the sample was incubated at 37 °C for at least one hour.
The mass spectrometry analysis of vip1 has been carried out using an Applied Biosystems (ABI, Foster City, California, U.S.A.) Proteomics Analyser 4800 MALDI-TOF/TOF device. The in silico digestion of vip1 and the calculation of the theoretical peptide masses, positions, and sequences has been performed by the PeptideMass tool of the ExPASy server (www.expasy.org/cgi-bin/peptide-mass.pl) [54,55].
3.6 Bioinformatic analyses
To search the Swiss-Prot database for potential homologues of vip1 and for related proteins, the Swiss Institute of Bioinformatics' (SIB) BLAST Network Service was used (ExPASy proteomics server, developed at SIB, NCBI BLAST 2 software) (www.expasy.org/). The pair wise alignment of the sequences of vip1 and the human Lissencephaly-1 protein was performed using the Needle programme (Matrix: Blosum62) of The European Molecular Biology Open Software Suite (EMBOSS) (http://www.ebi.ac.uk/Tools/emboss/align/index.html) [46]. For the three-dimensional structure of vip1, a request was sent to the SWISS-MODEL Server of The Expert Protein Analysis System (ExPASy) protein database (http://swissmodel.expasy.org) [47–49] and to the ModBase database of the UCSF (University of California, San Francisco) (http://modbase.compbio.ucsf.edu/modbase-cgi/index.cgi). The latter refers to the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB) for template proteins in order to create a structure model.
Acknowledgements
The authors thank the DFG (Deutsche Forschungsgemeinschaft), DFH/UFA (Deutsch-Französische Hochschule/Université Franco-Allemande) and MENESR (Ministère de l'Éducation nationale, de l'enseignement supérieur et de la recherche) (International Research Training Group 532 – Graduiertenkolleg GRK532) for financial support. We also thank the STEROLTALK project (Number: 512096) within the 6th Framework Program (European Union, EU) and Dr. Matthias Bureik for helpful discussions.