

1 Introduction
Hydantoin and its derivatives are biologically important compounds of relevance to chemists, biologists and pharmaceutical researchers. Over the last few decades, there has been considerable interest in the synthesis and characterization of hydantoin derivatives as an important class of heterocyclic compounds. Hydantoin derivatives that display interesting activities against a broad range of biological targets have been identified [1]. Hydantoins, substituted at C-5, are important medicinal compounds. Numerous applications have been found for hydantoin derivatives due to their antidepressant [2] and antiviral activities [3], the inhibition of binding of HIV to lymphocytes, [4] as well as their anticonvulsant and cardiac anti-arrhythmic effects [5]. Epilepsy is a group of chronic neurological disorders whose symptoms result from a brain dysfunction or an abnormal discharge of cerebral neurons. Drug therapy is the major treatment for epilepsy, and among the major drugs used in its treatment are the hydantoins [6]. The anticonvulsant properties of phenytoin (Dilantin®, Epilan®), one of the important drugs employed for the treatment of generalized convulsions, are responsible for the synthesis of many hydantoin analogs [7]. In the chemical industry, various 5,5-disubstituted hydantoins constitute the basis of a new generation of weatherproof high-temperature stable epoxy resins. Furthermore, hydantoin and hydantoin derivatives were used as a monomer for the synthesis of condensation polymers [8]. Hydantoin derivatives are synthetically valuable, e.g. as precursors to α-amino acid and pyruvic acid derivatives [9–11]. The structural characteristics of hydantoins and spirohydantoins are interesting in view of the development of novel drugs [12], and for better understanding the structure–activity relationship. As part of our research, the synthesis and characterization of series, new hydantoin-phosphonic acids and their analogues have been described [13–15]. The main goal of this study is synthesis of new hydantoin derivates as potential biologically active compounds. Many derivatives of these compounds have been synthesized and studied, but there are no data about the influence of the amino group and amino acids residue in the hydantoin ring on their biological activity. Based on already existed papers and our experience, we choose to synthesize some different derivatives of 5,5-dimethylhydantoin by TBTU/DIEA method.
2 Results and discussion
The synthesis and spectroscopic characteristics of many hydantoin derivatives are given in the literature [16–20], but no hydantoin consisting amino acid residue and consequently, their spectroscopic data are published up to now. In the present work, some different derivatives of 5,5-dimethylhydantoin were obtained in good yields by TBTU (2-(1-OH-benzotriazole-1-yl)1,1,3,3-tetramethyl-carbamide tetrafluoroborat)/DIEA (N,N-diiso propylethylamine) method. To prepare the new hydantoin derivatives (4a-4d and 5a-5c), we used, as a starting compound, 3-amino-5,5-dimethylimidazolidine-2,4-dione (2), whose synthesis was described in our previous work [15]. In order to synthesize compounds 4a-4d, we decided to use the corresponding protected amino acids, namely 3a-3d (Boc-Gly-OH, Boc-L-Leu-OH, Z-L-Phe-OH, Z-Gly-OH). Three novel dipeptides, 2-amino-N-(4,4-dimethyl-2,5-dioxoimidazolidin-1-yl) acetamide (5a), 2-amino-N-(4,4-dimethyl-2,5-dioxoimidazolidin-1-yl)-3-phenylpropanamide (5b), 2-amino-4-methyl-N-(4,4-dimethyl-2,5-dioxoimidazolidin-1-yl)pentanamide (5c), were synthesized by the deblocking of the Z-/Boc-group of the corresponding protected dipeptides. The corresponding protected dipeptides with Boc-group 4a and 4b were deblocked by Trifluoroacetic acid (TFA) for four hours at room temperature to obtain 5a and 5c, respectively. The corresponding protected dipeptides with Z-group 4c and 4d were deblocked by catalytic hydrogenolysis in the presence of Pd/C for five hours to obtain 5a and 5b, respectively. The synthetic pathway for the preparation of the novel compounds is given in the Scheme 1.
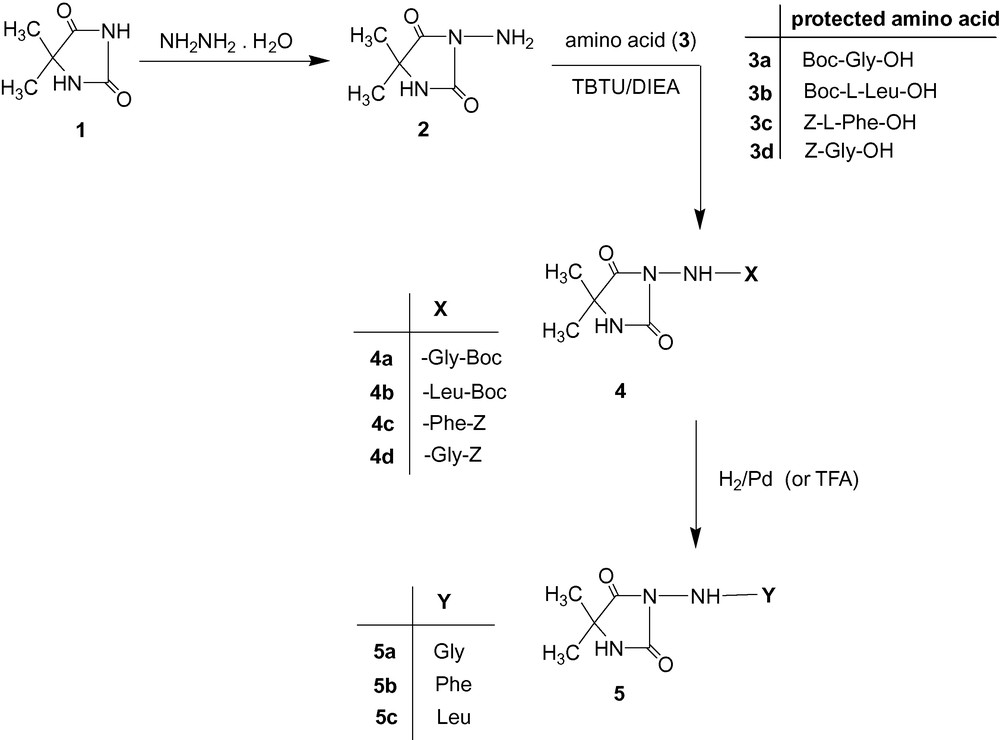
Synthesis of new hydantoin derivatives.
The newly synthesized compounds were characterised by melting points, thin layer chromatography (TLC), infrared (IR) and NMR spectroscopy, high performance liquid chromatography (HPLC) and MS (see Experimental section).
The IR spectroscopy study confirms the structure of the above-mentioned compounds. The IR spectra of hydantoin derivatives 5a-5c showed two strong peaks between 3200 and 3300 cm−1 that were assigned to the N-H of the imide and amide groups. Absorption bands between 1780 and 1730 cm−1 characterize the asymmetric and symmetric stretching of the carbonyl groups of the hydantoin. In the IR spectra of the compounds, 5a-5c absorption bands between 1690 and 1630 cm−1 (amide I) and 1580 and 1530 cm−1 (amide II) are observed which are characteristic peaks for amide groups.
The structures of all newly synthesized compounds have also been confirmed by 1H- and 13C-NMR spectroscopies. The singlet of 1H-NMR spectra of compounds 4a and 4b at 1.5 ppm can be assigned with the nine protons from the tert-butyl group. The singlet at 3.8 ppm and the triplet at 3.1 ppm for 4a and 4b, respectively, can be assigned to the proton from CH-group connected to NH-group. The signals of 1H-NMR spectra of compounds 4c and 4d at 4.7–4.8 ppm (singlet) can be assigned to the two protons from CH2 of Z-group and 6.8–7.7 ppm (multiplet) can be assigned with aromatic protons in Z-group. Signals for NH2-group at 4.8–4.9 ppm were observed in 1H-NMR spectra for all deblocked peptides 5a-5c.
Detailed spectroscopic data for all other hydantoin derivatives are presented in the Experimental section. As an example, we present here the 13C-NMR and MS spectra of compound benzyl 1-(4,4-dimethyl-2,5-dioxoimidazolidin-1-ylcarbamoyl)-2-phenylethyl carbamate (4c) (Fig. 1 and Fig. 2). The 13C-NMR spectrum of compound 4c, recorded in CD3OD solution, exhibits the four signals of carbon atom for C = O group, 155.16 (14C = O), 158.19 (2C = O), 173.11 (4C = O), 176.74 (5C = O) ppm. The signals in the diapason at 127–138 ppm in the 13C-NMR spectrum can be assigned to the carbon atoms of both aromatic systems (Fig. 1). In the 1H-NMR spectrum of this structure, there are signals at 4.40 ppm (triplet) that can be assigned to the CH proton, and the multiplet at 6.80-7.70 that can be assigned to the protons of two aromatic systems. Additionally in the same NMR spectrum, there is no signal at 4.8 ppm for the protons of the NH2 group characterizing the starting compound 2, which is a proof that the new compound 4c is obtained. These data from the NMR and MS spectra confirm the structure of the compound 4c.
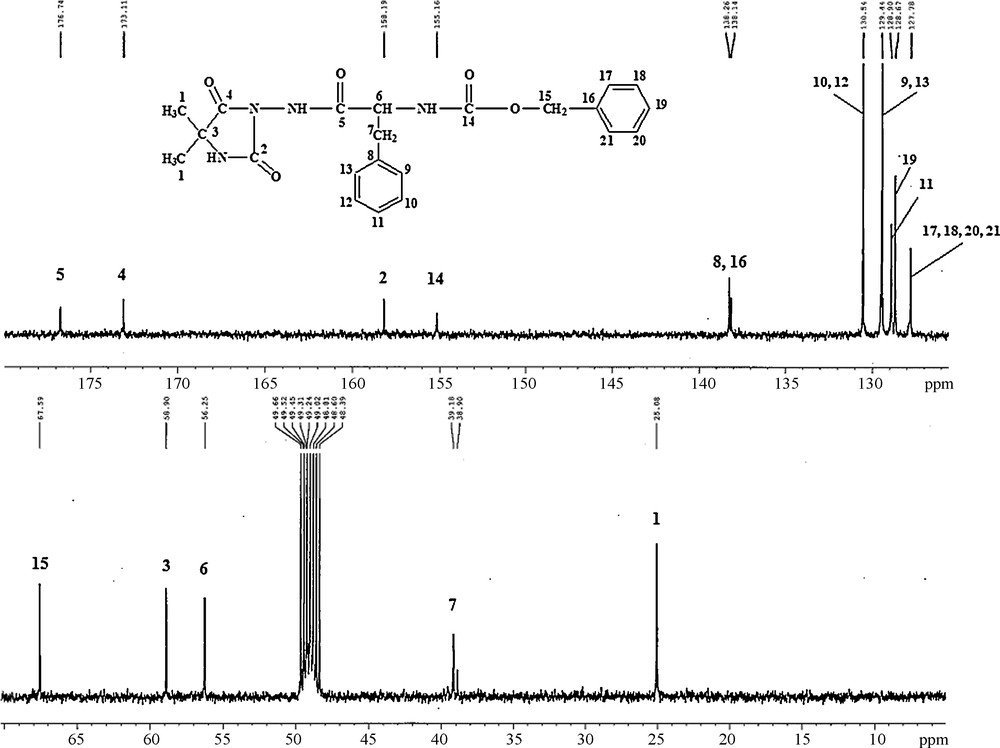
13C-NMR spectrum of benzyl 1-(4,4-dimethyl-2,5-dioxoimidazolidin-1-ylcarbamoyl)-2-phenylethylcarbamate (4c).

MS spectrum of compound 4c.
3 Conclusion
In this article, the synthesis and characterization of new hydantoin derivatives, prepared by reacting of 3-amino-5,5-dimethylimidazolidine-2,4-dione (2) and the corresponding protected amino acids (3a-3d) by TBTU/DIEA method, are described. After removing the Z-/Boc-group from the protected dipeptides, compounds (5a-5c) were obtained in good yields. The molecular structure of all compounds was studied by means of spectral methods. Further studies for the preparation and application of dipeptide derivatives with hydantoin moiety are currently in progress.
4 Experimental section
The protected amino acids were purchased from IrisBiotech (Germany). All other reagents and solvents were analytical or HPLC grade and were bought from Merck (Germany). The IR spectra were recorded in KBr pellets with a Perkin-Elmer Model 1600 Series FTIR instrument. Melting points (mp) were determined on a Koffler microscope and were uncorrected. Optical rotations were measured with a Perkin-Elmer 341 polarimeter. The purity of the products was checked by TLC on pre-coated plates of Silica gel 60 F254 (on recoated plates, Merck – Germany) using as mobile phase a 9:1 mixture of chloroform and methanol. Spots on TLS chromatograms were detected by chlorine/o-tolidine reaction and ninhidrine. The 1H and 13C-NMR spectra were obtained with a Bruker DRX 400 spectrometer at 400.13 and 100.61 MHz, respectively. 13C-NMR spectra were fully 1H decoupled. All chemical shifts were reported as δ values (ppm) relative to internal tetramethylsilane. The crude peptides were purified on a reversed-phase HPLC C18 column: flow 1 ml/min, H2O/CH3CN/0.1% TFA gradient 0 → 100% 20 min. The peptide purity was checked by electrospray ionization mass spectrometry.
4.1 3-Amino-5,5-dimethylimidazolidine-2,4-dione: 2
This compound was prepared according to the procedure described by us [15].
4.2 General procedure for preparation by the TBTU/DIEA (compounds 4a-4d)
3-Amino-5,5-dimethylimidazolidine-2,4-dione 2 (0.50 g, 1.00 mol), and the corresponding Boc-Gly-OH 3a (0.74 g, 1.20 mol), Boc-L-Leu-OH 3b (0.63 g, 1.20 mol), Z-L-Phe-OH 3c (0.76 g, 1.20 mol), and respectively, Z-Gly-OH 3d (0.88 g, 1.20 mol) were dissolved in Dimethylformamide (DMF) (4 ml). Then (1.20 mol) of TBTU (2-(1-OH-benzotriazole-1-yl)1,1,3,3-tetramethyl-carbamide tetrafluoroborat), (1.20 mol) HOBt (1-hydroxy benzotriazole) and (1.20 mol, ρ=0.76) of DIEA (N,N-diisopropylethylamine) were added. The reaction mixture was stirred at room temperature for 24 hours. After the end of the reaction, 10 ml water was added and the product was extracted by ethyl acetate (3 × 10 ml). The organic layer was washed with 5% NaHCO3 (3 × 10 ml), and water to pH 6–7 and dried over Na2SO4. The solvent was removed in vacuo and the crystallized product was purified by precipitation in ethyl acetate/petroleum ether.
4.3 Tert-butyl (4,4-dimethyl-2,5-dioxoimidazolidin-1-ylcarbamoyl) methylcarbamate: 4a
White solid, 80.5% yield; Rf = 0.55; mp 135–137 °C; 1H-NMR (400.13 MHz, CD3OD): δ (ppm): 1.55 (s, 9H, CH3), 1.71 (s, 6H, CH3), 3.83 (s, 2H, CH2-NH), 6.80 (s, 1H, NH), 7.95 (s, 1H, N-NH-CO); 13C{1H} NMR (100.61 MHz, CD3OD): δ (ppm): 21.90 (CH3), 28.65 (CH3), 52.10 (CH-N), 62.00 (-C-), 77.50 (-C-), 150.20 (2C = O), 156.07 (C = O), 165.04 (4C = O), 171.10 (C = O); IR (KBr, cm−1): 3314, 2980, 1793, 1750, 1682, 1530, 1157; ESI-MS: [M + 1] = 301.2; RP-HPLC: Rt 5.80 min.
4.4 Tert-butyl 1-(4,4-dimethyl-2,5-dioxoimidazolidin-1-ylcarbamoyl)-3-methylbutyl carbamate: 4b
White solid, 80.5% yield; Rf = 0.60; mp 115–118 °C; 1H-NMR (400.13 MHz, CD3OD): δ (ppm): 1.11 (d, 6H, CH3-CH), 1.24 (m, 1H, CH), 1.54 (s, 9H, CH3), 1.69 (s, 6H, CH3), 1.82 (t, 2H, CH2), 3.13 (t, 1H, CH-NH), 6.71 (s, 1H, NH), 8.60 (s, 1H, N-NH-CO); 13C{1H} NMR (100.61 MHz, CD3OD): δ (ppm): 21.06 (CH3), 22.50 (CH), 24.05 (CH3), 28.30 (CH3), 40.50 (CH2), 50.16 (CH-N), 61.07 (-C-), 79.50 (-C-), 148.20 (2C = O), 154.08 (C = O), 163.84 (4C = O), 168.90 (C = O); IR (KBr, cm−1): 3312, 2962, 2872, 1770, 1740, 1695, 1519, 1166; ESI-MS: [M + 1] = 357.2; RP-HPLC: Rt 6.50 min.
4.5 Benzyl 1-(4,4-dimethyl-2,5-dioxoimidazolidin-1-ylcarbamoyl)-2-phenylethyl carbamate: 4c
White solid, 86.0% yield; Rf = 0.68; mp 145–147 °C; 1H-NMR (400.13 MHz, CD3OD): δ (ppm): 1.70 (s, 6H, CH3), 3.15 (d, 2H, CH-CH2-Ar), 4.40 (t, 1H, CH-CH2), 4.81 (s, 2H, O-CH2-Ar), 6.80-7.70 (m, 10H, Ar), 8.80 (s, 1H, NH-CO); 13C{1H} NMR (100.61 MHz, CD3OD): δ (ppm): 25.03 (CH3), 39.18 (CH2-Ar), 56.25 (CH), 58.90 (-C-), 67.59 (O-CH2-Ar), 127-138 (C-Ar), 155.16 (C = O), 158.19 (2C = O), 173.11 (4C = O), 176.74 (C = O); IR (KBr, cm−1): 3300, 3063, 2981, 1790, 1740, 1695, 1521, 1258; ESI-MS: [M + 1] = 424.5; RP-HPLC: Rt 8.33 min.
4.6 Benzyl (4,4-dimethyl-2,5-dioxoimidazolidin-1-ylcarbamoyl)methylcarbamate: 4d
White solid, 79.0% yield; Rf = 0.65; mp 165–166 °C; 1H-NMR (400.13 MHz, CD3OD): δ (ppm): 1.71 (s, 6H, CH3), 3.80 (s, 2H, CH2-NH), 4.75 (s, 2H, O-CH2-Ar), 6.85-7.72 (m, 5H, Ar), 6.92 (s, 1H, NH), 8.15 (s, 1H, N-NH-CO); 13C{1H} NMR (100.61 MHz, CD3OD): δ (ppm): 22.45 (CH3), 42.10 (CH2-N), 60.59 (O-CH2-Ar), 65.60 (-C-), 126-137 (C-Ar), 151.20 (2C = O), 155.17 (C = O), 164.08 (4C = O), 172.00 (C = O); IR (KBr, cm−1): 3310, 3072, 2977, 1785, 1744, 1682, 1530, 1260; ESI-MS: [M + 1] = 335.2; RP-HPLC: Rt 6.65 min.
4.7 General procedure for deblocking of Boc-group. Obtaining of compounds 5a and 5c
The corresponding protected dipeptides 4a and 4b (1.00 mol) were dissolved in 10.00 mol TFA. The reaction mixture was stirred for four hours at room temperature. The end of the reaction was monitored by TLC. The excess of the solvent was removed in vacuo and the crystallized product was washed with cold diethyl ether.
4.8 2-Amino-N-(4,4-dimethyl-2,5-dioxoimidazolidin-1-yl)acetamide: 5a
White solid, 78.5% yield; mp 142–144 °C; 1H-NMR (400.13 MHz, CD3OD): δ (ppm): 1.69 (s, 6H, CH3), 3.73 (s, 2H, CH2-NH2), 4.8 (s, 2H, NH2), 6.7 (s, 1H, NH), 7.92 (s, 1H, N-NH-CO); 13C{1H} NMR (100.61 MHz, CD3OD): δ (ppm): 25.01 (CH3), 40.38 (CH2), 59.03 (-C-), 154.92 (2C = O), 167.14 (4C = O), 176.63 (C = O); IR (KBr, cm−1): 3300, 2982, 1773, 1736, 1681 and 1625 (amide I), 1575 and 1532 (amide II); ESI-MS: [M + 1] = 201.2; RP-HPLC: Rt 5.80 min.
4.9 2-Amino-4-methyl-N-(4,4-dimethyl-2,5-dioxoimidazolidin-1-yl)pentanamide: 5c
White solid, 70.2% yield; mp 135–136 °C; 1H-NMR (400.13 MHz, CD3OD): δ (ppm): 1.01 (d, 6H, CH3-CH), 1.21 (m, 1H, CH), 1.71 (s, 6H, CH3), 1.80 (t, 2H, CH2), 3.13 (t, 1H, CH-NH2), 4.90 (s, 2H, NH2), 6.90 (s, 1H, NH), 8.62 (s, 1H, N-NH-CO); 13C{1H} NMR (100.61 MHz, CD3OD): δ (ppm): 22.00 (CH3), 23.50 (CH), 23.85 (CH3), 40.50 (CH2), 49.66 (CH-N), 60.11 (-C-), 148.20 (2C = O), 163.84 (4C = O), 168.90 (C = O); IR (KBr, cm−1): 3315, 2960, 2881, 1772, 1732, 1678 and 1639 (amide I), 1575 and 1541 (amide II); ESI-MS: [M + 1] = 257.3; RP-HPLC: Rt 5.78 min.
4.10 General procedure for deblocking of Z-group by catalytic hydrogenolysis in the presence of Pd/C. Obtaining of compounds 5a and 5b
The corresponding protected dipeptides 4c and 4d (1.00 mol) were dissolved in 8 ml MeOH and then, Pd/C and 0.1 ml of 25% HCl were added. Hydrogen was passed through the reaction mixture at room temperature for five hours. The deblocking of the protecting groups was watched on TLC and, after finishing the reaction, Pd/C was filtered out and MeOH was evaporated in vacuo.
4.11 2-Amino-N-(4,4-dimethyl-2,5-dioxoimidazolidin-1-yl)-3-phenylpropanamide: 5b
White solid, 75.0% yield; mp 175–177 °C; [α]20D = + 59.4 (c 0.1, MeOH); 1H-NMR (400.13 MHz, CD3OD): δ (ppm): 1.68 (s, 6H, CH3), 3.20 (d, 2H, CH-CH2-Ar), 4.46 (t, 1H, CH-NH2), 4.87 (s, 2H, NH2), 6.75 - 7.81 (m, 5H, Ar), 7.90 (s, 1H, N-NH-CO); 13C{1H} NMR (100.61 MHz, CD3OD): δ (ppm): 25.10 (CH3), 39.22 (CH2-Ar), 57.05 (CH), 59.00 (-C-), 127-138 (C-Ar), 158.19 (2C = O), 173.11 (4C = O), 176.74 (C = O); IR (KBr, cm−1): 3305, 2985, 2890, 1774, 1741, 1681 and 1636 (amide I), 1580 and 1533 (amide II); ESI-MS: [M + 1] = 291.2; RP-HPLC: Rt 7.50 min.
Acknowledgement
We gratefully acknowledge the financial support by Bulgarian National Fund of Scientific Research and the University of Chemical Technology and Metallurgy – Sofia – contract № 10646.