

1 Introduction: catalysis and green chemistry
One critical aspect of the emerging “green chemistry” field is catalysis [1]. Most chemical reactions are indeed too slow and too unselective under normal conditions to have direct industrial applications. This is the consequence of the existence of several chemical pathways with large and comparable activation barriers. The kinetic theory suggests that these pathways can be accelerated to large extents at high temperatures and indeed most industrial uncatalyzed synthetic processes require heating, however at the expense of selectivity so that a given product is obtained with low yields. This obviously introduces an extra energy cost and most often negative impacts on the environment through the consumption of fossil fuels, the emission of carbon dioxide, a greenhouse effect gas, and the production of significant quantities of waste (solvents, by-products…) which have to be reprocessed.
Catalysis provides a solution to these issues in many cases. Indeed a catalyst is a chemical, a small soluble molecule, a macromolecule or a solid material, which selectively and transiently interacts with one or several reactants and translates this interaction into a large decrease of the activation barrier of one of the possible pathways. As a consequence good yields of a unique product can be obtained at fast rates and relatively low temperatures.
A second very important property of a catalyst is that, whereas it can be transiently modified in the course of the reaction, it is recovered in a free fully intact and active state once the products of the reaction have been released. This has an important implication, namely that a given amount of the catalytic compound can be used for transforming a much larger amount of substrate molecules (actually an infinite amount in the case of an ideal catalyst that would never get inactivated) and, as a consequence, a small quantity of catalyst is needed. Such a catalytic activity is thus defined in terms of turnover numbers (the ratio between the number of transformed reactant molecules and the number of catalyst molecules present in the reaction mixture). Catalytic processes are consequently energy-saving, much more economical and more environmentally friendly than stoichiometric processes, as they generate much less waste.
This explains why, during the last 50 years, the chemical and pharmaceutical industry has constantly introduced catalysis into synthetic reactions. Research of new, effective and selective catalysts is currently one of the major industrial approaches in chemistry, resulting in the development of modern and sustainable methods for making cheap materials for the sake of human health and well-being. As a matter of fact approximately 80% of the products made by the chemical and pharmaceutical industry involve one or several catalytic steps during synthesis [2].
2 Biocatalysis
The question is then how to find these superb catalysts. One increasingly developed approach is based on the exploitation of living organisms. Indeed the most impressive catalysts on earth are found in Nature. Enzymes, the name for biological catalysts, clearly constitute one of the key secrets of life. There is no life without a huge capacity for accelerating, in a controlled way, the thousands of cellular reactions and for making these reactions highly chemo-, regio- and stereoselective, at the cheapest energetic price. Enzymes are those fascinating proteins, which are responsible for the extraordinary rates and selectivities of biological reactions, still unapproachable in chemical synthesis laboratories [3]. This explains why an increasing use of biocatalysis in synthesis is expected to dramatically impact the production of chemical compounds, drugs and materials [4]. Whereas biotechnological synthetic processes currently represent only 5% of the chemical industry market, it is expected that this number will be much closer to 30% by 2020 [5]. Indeed these processes are likely to provide high-energy savings and low environmental negative impacts. As an example, the BASF company now produces 1000 tons of vitamin B2 per year, which is mainly used for animal feeding, through a biotechnological process based on the utilization of filamentous fungi as a synthetic factory. The single-step process has replaced the standard chemical process, with eight chemical reaction steps, resulting in a remarkable economy of 40% on the product cost, 30% on CO2 emissions and 95% on waste [6].
Synthetic biotechnology is not restricted to the use of intact wild-type living microorganisms (bacteria, fungi, yeast…). Identification and cloning of the gene coding for an enzyme catalyzing a reaction of interest can result in its exploitation in synthetic processes. The latter are thus based on the utilization of either a bacterial strain overexpressing the corresponding gene or the purified, often recombinant, molecular enzyme. In general, a living factory, i.e. a cell, is preferred because pure enzymes are trickier to use in industrial applications: they require complex and expensive purification procedures and are quickly inactivated during catalysis. In addition, a major limitation of both approaches resides in the reaction conditions under which a synthetic process can be carried out: in general ambient temperature and pressure, use of water as the only solvent, neutral pH… Future strategies using living organisms and pure enzymes will take advantage of the exploration of the biodiversity. Indeed we currently know less than 1% of the Earth's microbial world [7] and discovery of new bacterial strains, living under extreme conditions (temperature, pressure, salt, pH…) and containing amazingly stable and efficient enzymes, are expected to result from this exploratory work, thus providing a possible broadening of the range of conditions that can be used in biotechnological processes.
3 From biology to new catalysts: synthetic biology and bioinspired catalysis
The utilization of microorganisms and enzymes as catalysts has two main drawbacks. The first one, mentioned in the above paragraph, is related to the complexity of the catalytic system, which makes it expensive, fragile and active only under very narrow reaction conditions. One solution in our opinion resides in the bioinspired chemistry approach that we extensively discuss below. Imitating the active site of an enzyme in the form of a small synthetic chemical material allows one to reduce the complexity. The second drawback resides in the extreme selectivity of enzymes. This seems to be a rather provocative statement but, indeed, this selectivity implies that only a limited number of compounds, the natural substrates, can be transformed to produce the natural products, exclusively. Bioinspired catalysis provides a way to broaden the scope of possible substrates since the enzymatic active sites’ analogues are more substrate-accessible and flexible [8]. Another emerging approach to address this issue is synthetic biology. Synthetic biology is discussed extensively in other chapters of the present journal issue and will be only briefly defined here to show that synthetic biology and bioinspired catalysis are two possible approaches, very different in the methods but with similar objectives, expanding natural processes towards the use of unnatural substrates and their transformation into unnatural materials, with fascinating possible applications for chemical industry.
3.1 Synthetic biology
As discussed above, a biotechnological process is currently based mainly on the utilization of a single enzyme catalyst or on a bacterial strain overexpressing a single gene. Synthetic biology is based on an extreme exploitation of genomes and molecular biology technologies [9,10]. It aims at engineering new biological pathways in cells and at creating wholly new organisms from scratch. This is expected to generate organisms containing entire non-native pathways and organisms with completely new genomes. This unnatural expansion of life, which requires new technologies for the transfer of multiple genes into cells in controllable ways, raises serious biosafety concerns. However, if this aspect is under control, it might not only provide a fantastic tool to understand, at the very fundamental level, what life is, but also open new possibilities for biotechnological production of useful new products, drugs, biofuels and new types of enzymes.
3.2 Bioinspired catalysis
A probably safer and simpler strategy for expanding the repertoire of “biocatalysts” is bioinspired catalysis. Here the approach implies that the increased understanding, at the very molecular level, of a given biological catalyst is exploited not only to reproduce it synthetically in a much simpler form (biomimetic chemistry), but also to explore unnatural combinations of atoms and molecules, that organisms have neither experienced nor kept during evolution for multiple reasons (bioavailability, selection pressure, toxicity…) to generate original catalysts (bioinspired chemistry) for the same function. The variations introduced in the latter case are expected to improve the catalytic efficiency of the simplest biomimetic model and expand the range of substrates that can be transformed as well as the types of reactions catalyzed. Obviously, the imitation and its variations should not be limited to the structure of the active site but rather essentially directed towards its reactivity [11].
It is important to understand the difference between the restricted biomimetic chemistry and the open bioinspired chemistry. In the first case, the chemist is driven by the fascinating possibility to synthesize from scratch a piece, even a small part, of a living system with identical functional, structural and spectroscopic properties. The bioinspired approach does not aim at imitating/replicating the exact details of an active site but instead exploits its basic chemical principles, on which a much larger chemical space can be explored, to enrich chemistry with new catalysts and new exciting reactions. The flow of information from biology to chemistry is much larger in the second case. A repeatedly reported symbolic example of a bioinspired object is a jumbo jet which flies as a bird but is not just a scaled-up bird [12]. In the following we present an interesting example, more modest than the one that led from a bird to a plane, of a bioinspired approach developed in our laboratory which resulted in a remarkable catalytic material for water to hydrogen interconversion with potential technological applications in renewable energy systems.
4 From hydrogenases to bioinspired nanocatalysts for H2 oxidation/production
The development of a bioinspired homogeneous metal catalyst is a multidisciplinary approach which implies the following steps: (i) detailed characterization of the selected metalloenzyme and of its active site (structure and mechanism); (ii) bio-inspired design of the coordination/organometallic compound through the understanding of the structural/chemical principles on which the active metal site is built and which were identified during the first step; (iii) synthesis of new ligands and their coordination to metal centres so as to assemble the soluble low-molecular weight metal complexes (organic and inorganic synthesis); (iv) finally, evaluation of their catalytic performances for a given reaction. Such catalysts have a number of advantages. They are generally designed to be easy to synthesize and characterize; they are cheap to produce on a large scale if one succeeds to avoid noble and expensive metals such as Pt, Pd, Ir, Rh, Ru or Au; they can be used in different organic solvents and some of them are also water-soluble; they can function under very different temperature and pressure conditions. On the other hand, a general assumption is that such compounds suffer from much lower catalytic rates and turnover numbers than those of enzymes and generally display poor selectivities. Another important issue to consider is catalyst recycling. When the system is homogeneous this is a difficult process and, as a consequence, when an efficient soluble catalyst is discovered, appearing as a good candidate for technological applications, it is more and more requested that this compound be evaluated functionally also in an insoluble form, after heterogeneization procedures involving covalent or non covalent attachment to the surface of a solid material. Whether this improves or damages the catalyst performance cannot be anticipated in general.
The reaction discussed here, in order to illustrate the bioinspired approach discussed above, is the reversible interconversion of protons and hydrogen (Eq. (1)).
| (1) |
This reaction is becoming important in the new energetic context of the planet because H2 can be considered as a unique energy vector. Indeed transformation of water to H2, through electrolysis or photolysis, is an efficient way to store the energy (electricity or sunlight) in a stable chemical form and oxidation of H2 to water using atmospheric oxygen is a simple way to recover the energetic input, with no production of greenhouse gases such as CO2, and no formation of atmospheric pollutants such as NOx or ozone [13,14]. However, these reactions are complex multielectronic processes which require catalysts for being practically useful and an intense effort of research is spent all over the world to find a solution to this catalytic problem. Indeed, even though rather efficient, still too expensive, available technological devices, such as electrolyzers (for conversion of electrical energy to chemical energy through H2 production) and fuel cells (for conversion of chemical energy to electrical energy through H2 oxidation), in most cases integrate unsustainable and expensive metals such as platinum for catalysis. Simple calculations demonstrate that there is no future for cars equipped with H2-fuel cells just because there is not enough Pt at the surface of the Earth to sustain their long-term production [15]. In addition, such a widespread utilization of fuel cells for transportation will result in the dissemination of significant amount of platinum in the environment because of the unavoidable dissolution of the catalyst during catalysis and its elimination as a solute in the exhausted water. Thus, aside for public health issues related to this toxic heavy metal (same currently applies for catalysts used in catalytic converters for automobiles), it is currently not possible to recycle more than 50% of the platinum contained in a fuel cell. Imagine the 700 million of cars present on earth equipped with such devices: within 15 years, all our Pt resources will be consumed. This is the reason why an intense current line of research is in the discovery of original catalysts using first-row transition metals such as nickel (Ni), cobalt (Co) and above all iron (Fe), one of least expensive and the most abundant on the Earth.
Characterization of certain living organisms, such as archaea, bacteria, cyanobacteria and algae, has led to the exciting discovery that both reactions of Eq. (1) are carried out very efficiently (high turnover frequencies) at moderate temperatures and pressures within living cells. For example, bacteria such as Ralstonia eutropha (a facultatively chemolithoautotrophic organism) are able to use hydrogen as the sole energy source and have developed a metabolism based on the reaction of hydrogen with oxygen [16], the same that which operates in fuel cells! Another example comes for micro-algaea such as Chlamydomonas reinhardtii, able under some specific conditions to use sunlight to drive the reverse reaction, i.e. splitting water into hydrogen and oxygen [17]. This chemical activity is made possible through the expression of fascinating metalloenzymes called hydrogenases [16,18], which catalyze these reactions without any overvoltage (the extra energy required for the reaction over that defined by the standard potential of the redox H+/H2 couple; in other words, the difference between the potential that needs to be applied to the system to allow it to function and the standard potential of the redox H+/H2 couple) and at very high rates (one molecule of hydrogenase produces 1500 to 9000 molecules of H2 per second at pH 7 and 37 °C in water) [19,20]. When careful examination of these enzymes, including through high resolution X-ray crystallography at the end of the 1990s [21–24], revealed that the metals in action were non precious metals such as nickel and iron, it became obvious that a solution to the catalytic problem of Eq. (1) could be found in Nature [14]. Practically, again, this conclusion was translated into the development of the three following classes of bio- and bioinspired catalysts: (i) the microorganisms themselves, with a careful selection of the most productive natural or genetically modified ones; (ii) the purified, often recombinant, hydrogenases; (iii) the biomimetic/bioinspired compounds. In our laboratory, we have pursued the third strategy which is described in the following chapter.
4.1 Hydrogenases
The metallo-enzymes responsible for hydrogen metabolism in a number of microorganisms are called hydrogenases [25]. These enzymes can catalyze both hydrogen uptake, furnishing the cell with low-potential electrons, and hydrogen evolution, which re-oxidizes the pool of electron carriers participating in fermentation or photosynthesis. There are two classes of hydrogen-metabolizing enzymes: the [NiFe]- and [FeFe]-hydrogenases. They have catalytic dinuclear metal centres (Fig. 1) in which either one nickel and one iron or two iron atoms are connected through two thiolate bridges. The bridging thiolates have different origins in the two enzymes. In [NiFe]-hydrogenases, they are provided by two cysteine residues, so that Ni is coordinated by four cysteinates in a non-regular configuration. In [FeFe]-hydrogenases the bridge is made from a small organic dithiolate ligand. X-ray crystallography and FTIR spectroscopy have clearly shown that the two kinds of hydrogenase are characterized by active sites that contain cyanide (CN–) and carbon monoxide (CO) molecules as ligands of an iron centre. Besides the curiosity for the presence of such toxic molecules as constituents of a native enzyme, these common Fe(CN)y(CO)x units make hydrogenases some of the extremely rare examples of organometallic centres in biology. The structures of the metal sites of [NiFe]- and [FeFe]-hydrogenases, derived from detailed crystallographic studies, are shown in Fig. 1. In the case of the [FeFe]-hydrogenase active site, the bridging ligand has been very early proposed to be a dithiomethylamine molecule [26] and this was recently confirmed from spectroscopic studies [27]. It is assumed that the amine basic group plays a very critical function as it participates in proton exchange, facilitating proton reduction to H2 or proton uptake from H2. The proton-exchange site in [NiFe]-hydrogenases is less clear, but is likely to reside on the terminal Ni-bound cysteinate residues [28].

Schematized structures of reduced forms of the [NiFe]-hydrogenases (left) and the [FeFe]-hydrogenases (right, X is a hydride ion, dihydrogen or a water molecule).
Finally, what the three-dimensional structures have also revealed is the presence of an array of iron-sulfur clusters, distant from each other by less than 10 Å, allowing an electrical communication between the active site and the protein surface where redox partners are expected to bind for accepting (H2 oxidation) or providing (H2 formation) electrons (Fig. 1). At this surface, a so-called distal cluster is exposed to solvent and plays a crucial role in connecting the hydrogenase to its redox partners. A remarkable property of these enzymes is that they can function, in pure isolated form, as excellent electrocatalysts for H2 oxidation or H+ reduction when adsorbed on a carbon electrode surface with overvoltages and current densities rivaling those of Pt [20].
These enzymes are unfortunately not easy to use in biotechnological applications. First, they are multisubunit proteins present in small amounts in microorganisms and their production is not easy. Second, they are, in general, extremely sensitive to oxygen and thus can only be handled under anaerobic conditions, which is highly prohibitive for technological applications. Third, they contain unique cofactors, described above, which require complex and largely unknown protein machineries for maturation [29–33] thus making production of recombinant enzymes very difficult. Utilization of hydrogenases in electrolyzers or fuel cells will thus require a huge amount of work, even though interesting results using slightly air-resistant enzymes have been recently reported by F. Armstrong's group [34–36]. This suggests that there is a need to better and more extensively explore the biodiversity in order to discover hydrogenases with greater stabilities.
4.2 Hydrogenase model compounds
Starting from the very detailed structural information summarized above a number of chemists succeeded in synthesizing original complexes which nicely imitated the active sites of the hydrogenases, through the bioinspired approach [37,38]. In Figs. 2 and 3 are displayed structures of some examples of dinuclear Ni-Fe and Fe-Fe model compounds to show how close synthetic complexes can reproduce the structural features of an enzyme active site. The common themes for Ni-Fe compounds are: (i) a nickel ion tetracoordinated with four sulfur ligands, two of them bridging the two metal ions; (ii) an organometallic Fe centre (preferentially with CO and CN– ligands but phosphine can be used as a surrogate for cyanide). The common themes for Fe-Fe compounds are: (i) two organometallic Fe centres (preferentially with CO and CN– ligands); (ii) a small dithiolate bridge. Very recently, a number of excellent articles have reviewed the impressive biomimetic work achieved during the last 10 years and readers are invited to refer to them [37,38]. They demonstrate the unique potential of chemistry not only to strictly imitate Nature but also to derive from it some original analogues, allowing visiting a much larger chemical repertoire than Nature does. More than 50 different dinuclear Ni-Fe biomimetic complexes have been reported in the literature since 1996. A very large number of Fe-Fe biomimetic complexes has been shown to display electrocatalytic activity, however exclusively for proton reduction to H2 and with large overvoltages. In contrast, surprisingly, none of the Ni-Fe mimics was found to be active as an electrocatalyst for H2 production or oxidation until, in 2009, Barton et al. reported on the protonation of a Ni(I)-Fe(I) compound, with a P2S2 set around the nickel (Fig. 4), yielding a stable bridging hydride product and showed that the latter was competent for catalyzing hydrogen evolution from trifluoroacetic acid in CH2Cl2 [62], however, with high overvoltages.
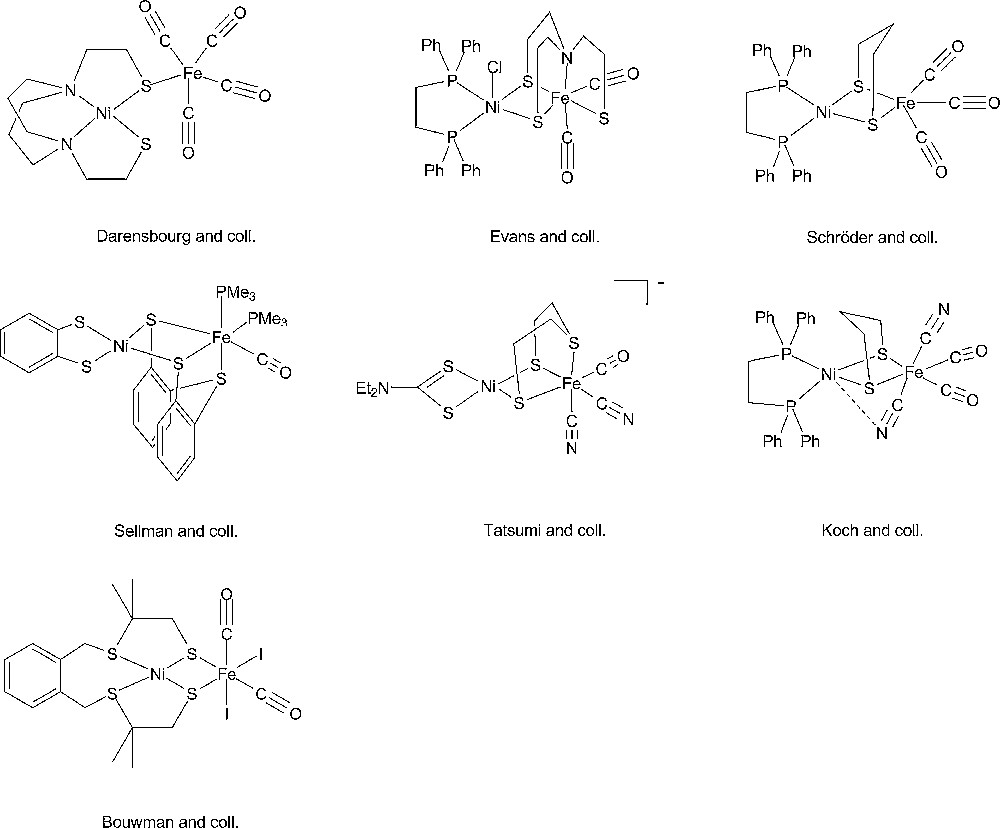
Selection of representative examples of model dinuclear Ni-Fe complexes from the groups of Darensbourg [39], Evans [40], Schröder [41], Sellmann [42], Tatsumi [43], Koch [44] and Bouwman [45].
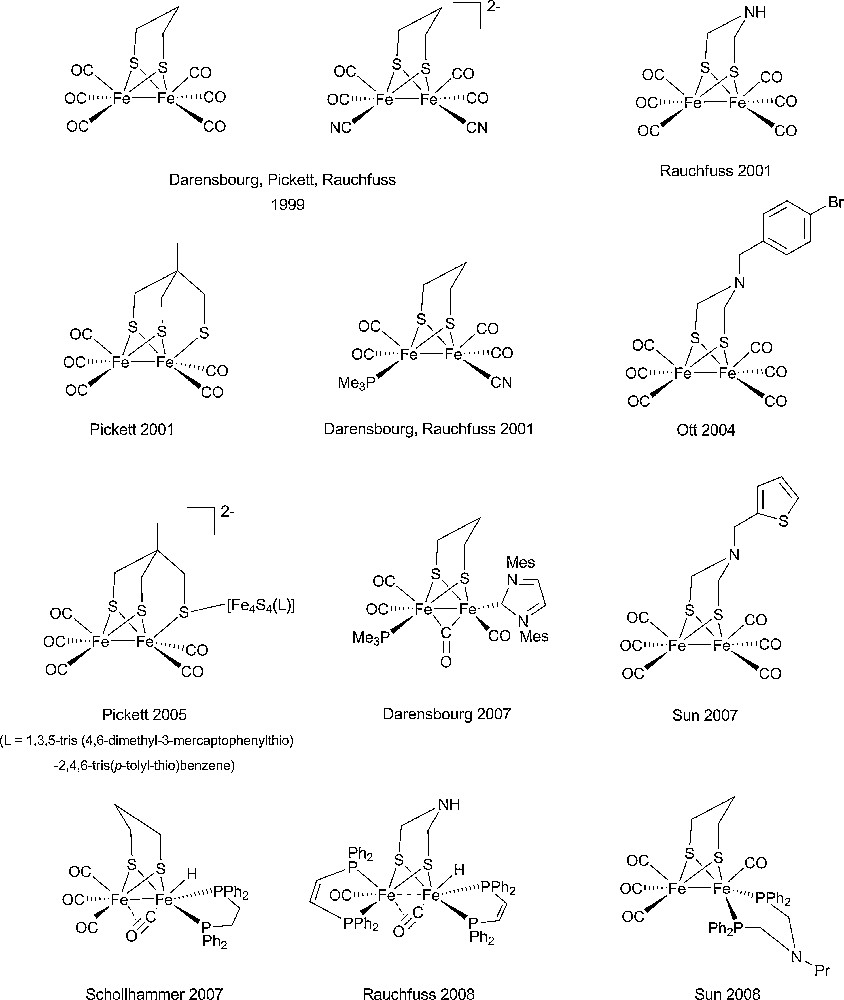
Selection of representative examples of model dinuclear Fe-Fe complexes from the groups of Pickett [46–48], Rauchfuss [49–52], Darensbourg [53–57], Sun [58–60], Ott [59] and Schollhammer [61].
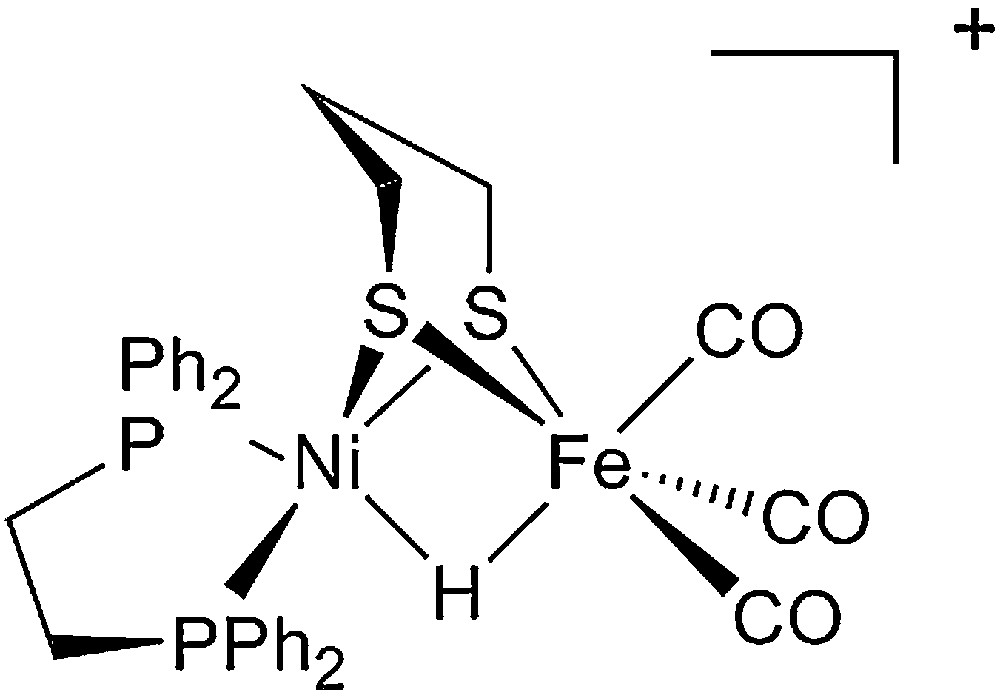
Structure of the functional Ni-Fe biomimetic model of [NiFe]-hydrogenases reported by Rauchfuss et al. [62].
Another, now more bioinspired than biomimetic, approach to extend this repertoire was developed by the group of Ogo [63] and ourselves [64–68] during the last years through the synthesis and the study of interesting dinuclear Ni-Ru complexes (Fig. 5). Unfortunately, so far, these complexes function only in organic solvents (DMF, CH2Cl2, CH3CN) with overvoltages in the 0.5-1 V range, thus excluding their exploitation in technological applications.
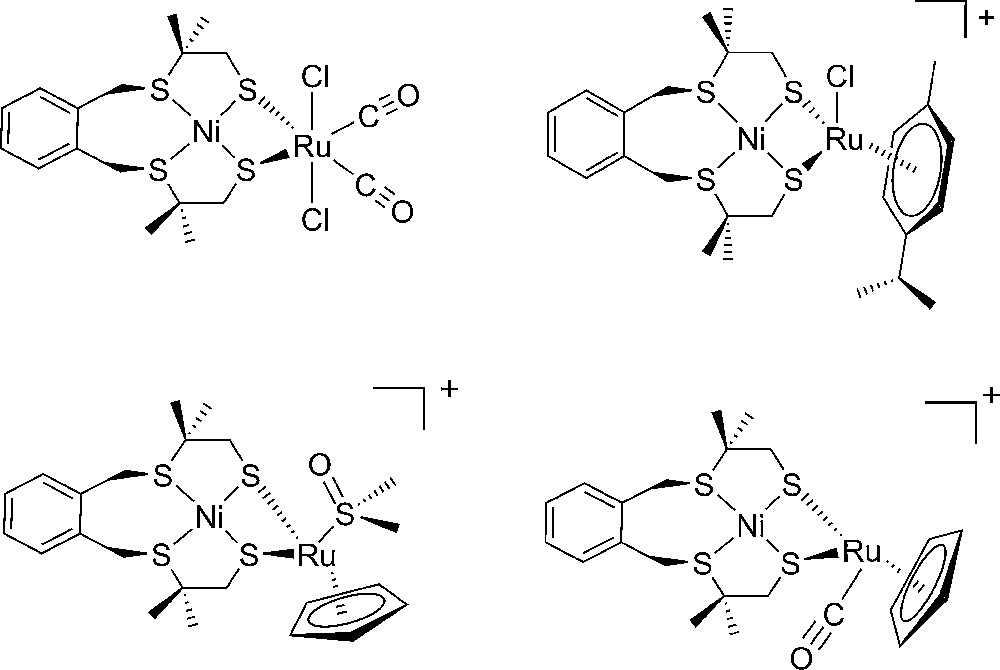
Structure of some functional Ni-Ru bio-inspired models of [NiFe]-hydrogenases.
It is tempting to conclude from this important piece of work that the bioinspired approach, even though it is successful in structurally imitating the first coordination sphere of a biological metal centre, is too simplistic to reproduce the fine functional properties of an enzyme. In this particular case, it fails, for example, in generating catalysts working at the standard potential of the H2/H+ redox couple, as the enzyme does. This is not fully surprising since an enzyme, such as hydrogenase, is not restricted to its active site. Its whole complex molecular machinery contains a polypeptide polymer which provides a unique medium to the active site, channels for H2 transport as well as a scaffold on which various critical functionalities, such a proton exchange sites and electron-transfer centres, are optimally distributed, an arrangement which results from billions of years evolution. Should chemists then try to imitate the whole protein or give up? Neither. On the one hand, it seems absurd to try to copy such a whole complex enzyme. This is just impossible, would be too expensive and time-consuming and in full contradiction with the finality of the biomimetic/bioinspired chemistry which basically aims at producing simple, but still active, and cheap forms of the enzyme, even if it is accepted that the simplification is paid by a certain loss of activity. On the other hand, there is a need to better understand, at the molecular level, what makes this particular enzyme so selective and so active and the tremendous synthetic power of chemistry allows introducing some, albeit limited, complexity in the bioinspired architectures to improve their functional properties significantly. The example below provides one illustration that this is a viable strategy.
The exquisite work by D. L. DuBois [69–71], recently reviewed [72,73], has shown that active complexes could be produced by combining features of both [NiFe]- and [FeFe]-hydrogenases. Dubois mononuclear nickel diphosphine complexes (Fig. 6) are structurally rather distant from hydrogenase active sites (Fig. 1). From the [NiFe]-hydrogenases, they borrow the Ni ion and from the [FeFe]-hydrogenases they take the pendant proximal base, an amine group, but as part of a diphosphine ligand instead of the dithiolate ligand found at the enzymatic active site. These compounds have been shown to display remarkable catalytic properties. Depending on the nature of the substituents at the N and P atoms and the acidobasic conditions of the medium, they catalyze either the electroreduction of protons (in the form of triflic acid or protonated DMF) to hydrogen or the electrooxidation of hydrogen in the presence of triethylamine with relatively small overvoltages, in the range of 0.1–0.2 V. Correct positioning of the basic amine centres close to the nickel atom facilitates intra- and intermolecular proton transfers and the coupling of proton and electron transfer processes, thus resulting in a dramatic decrease of the overvoltage. This is also the only class of molecular catalysts with members reported to electrocatalyze H2 oxidation with such low overvoltages. The importance of a basic site in the second coordination sphere has also been shown recently in the case of mononuclear cobalt complexes either with the same diphosphine ligands [74] or with original diimine-dioxime ligands providing an open oxime bridge susceptible to proton exchange (Fig. 7) [75]. These complexes are indeed excellent electrocatalyts for proton reduction and require small overvoltages. These examples nicely demonstrate that increased understanding of the chemical principles on which the reactivity of a biological active site and fine utilization of the synthetic power of chemistry allow minor modifications of a bioinspired catalyst resulting in considerable functional improvements.

Structure and reactivity of DuBois catalysts for homogeneous hydrogen evolution or uptake [70,71].

Structure of cobalt catalysts for hydrogen evolution with proton relays in the second coordination sphere from D.L. DuBois (left) [74] and our group (right) [75].
4.3 Towards electrocatalytic materials
The last step of this story has been achieved in our laboratory and has been recently published [76]. Further exploitation of DuBois complexes was considered when we decided to evaluate the potential of hydrogenase-bioinspired catalysts as heterogeneized electrode materials for applications in electrolyzers or fuel cells. This led us to synthesize an analogue of DuBois complexes, namely 1(BF4)2, in which a functionality, an activated ester, was introduced to allow a covalent attachment, through the formation of an amide bond, to a carbon nanotube decorated with amine groups at its surface (Fig. 8).

Electrografting of amine groups on MWCNTs and postfunctionalization with complex 1(BF4)2 through amide links. This is a simplified representation of the structure of the material for sake of clarity: the number of phenylene residues is indeed arbitrary and attachment of the nickel complex to two or more surface amine groups not excluded.
The latter (Fig. 8) were produced during electrochemical one-electron reduction of the 4-(2-aminoethyl)phenyldiazonium salt 2(BF4)2 since this reaction generates N2 and aryl radicals which react with the carbon surface to form covalent CC bonds between the grafted group and the carbon nanotube (Fig. 8). It should be mentioned that carbon nanotubes were chosen not only and reactivity of DuBois catalysts for homogeneousfor their outstanding electron conductivity, but also because the nanostructuration of their films provides a large surface area from which one can expect high catalyst loading. Deposition of a thin film of these electroactive Ni-functionalized carbon nanotubes onto a carbon substrate generated a cheap, stable, air-resistant cathode material with remarkably unique performances especially under the strongly acidic conditions required in the expanding proton exchange membrane (PEM) technology. Hydrogen indeed evolves from aqueous sulphuric acid solution with very low overvoltages (<20 mV) and exceptional stability (> 100000 turnovers). Interestingly, this Pt-free catalyst is also very efficient for hydrogen oxidation under the same conditions with current densities similar to those observed for hydrogenase-based materials (1–5 mA.cm-2). The latter property was completely unexpected from solution studies and showed how modification of the distant environment of a metal complex, let us say its third coordination sphere, may also allow us to tune its catalytic properties. As a result of this finding and to the best of our knowledge, this has been the first report of a molecular-engineered and noble-metal free electrode material that is capable to achieve hydrogen evolution/oxidation with no or little overvoltage (Fig. 9).

Schematic representation of the structure and reactivity of the bioinspired nickel catalyst grafted on a carbon nanotube. Electrons are exchanged between the carbon nanotube and the Ni centres where H+ is reduced to H2 or H2 oxidized to H+.
5 Conclusion
An unexpected conclusion of this work is that the attachment of the catalyst to carbon nanotubes has greatly improved its stability and thus its activity in both reactions. It is still unclear how grafting onto carbon nanotubes contributes to this amelioration, even though it is likely that their remarkable electronic conductivity, which probably optimizes the electrical connection between the catalyst and the electrode, plays an important role. It is exciting to suggest that, in some way, the conducting carbon nanotubes imitate the electrical linear circuit provided by the electron-transferring iron-sulfur clusters, which in the enzymes connect the buried active site to the surface of the protein (Fig. 1). It would be incorrect to say that the use of carbon nanotubes was inspired by this biological intramolecular electrical pathway during the design of the Ni-based electrode material. Nevertheless, it remains that this material is a beautiful truly bioinspired chemical object, a new and simpler version of an hydrogenase, containing a non-noble metal, nickel, for H2 formation and activation, basic sites, the amine groups, for proton exchange, and an electron-transferring device, the carbon nanotube, for electron exchange. In terms of mimicking complex biological systems, it is a modest contribution, but it is the type of things that chemists can reasonably achieve. In terms of potential applications, because of the great stability and the excellent activity of this original material, it opens very exciting and promising perspectives for the new technologies of energy, more exciting and promising than those opened by the utilization of fragile and expensive hydrogenases, so far. This is a good demonstration of how bioinspired chemistry can play a major role in the development of new technological breakthroughs.