

1 Introduction
Compared to alternative electrochemical energy storage technologies, Li-ion batteries offer the best promises for many applications, because of their larger energy density, larger power density, and larger long-term duration. However, new coming applications are high performance-demanding such as clean personal transportation (Electric Vehicles [EVs], Hybrid Electric Vehicles [HEVs], Plug-in Hybrid Electric Vehicles [PHEVs], Light Electric Vehicles [LEVs]), power tools and storage of intermittent renewable energies. For these applications, besides safety, low cost, and environmental friendliness, more energy density, more power density, or longer life (shelf life or cycle life) are greatly required. These energy, power and life issues are closely related to material and interfacial ones.
In order to achieve energy, power and life performance, most research efforts of the last 20 years have been devoted to the following aspects: (i) new structures and new compositions of the electrochemically active material (AM), engineering of their particle size, morphology, surface coatings, and better understanding of their electrochemical behavior through novel or in situ characterization techniques; (ii) new lithium electrolytes and characterization of the solid–electrolyte interface (SEI), (iii) new reaction mechanisms (e.g., conversion and displacement reactions). In contrast, the integration of AM within the electrode, i.e., the composite electrode engineering or – in other words – the formulation of the composite electrode, has received only little attention by the academic community so far.
The positive or negative electrode of a lithium-ion battery (LIB) is in fact a composite material that needs to bring efficiently the ionic reactants and the electrons to the surface of the electrochemically AM particles. This composite material needs to possess mixed conductivity with both Li+ ionic and electronic conductivity. Such a complex medium is generally obtained by mixing together the AM grains with non-electroactive additives such as a very fine powder of electronically conductive carbon (C) and a binder (B). The carbon additive is supposed to ensure electronic percolation within the composite electrode and to improve the AM particle-to-particle electronic contacting. The latter is known to be a major source of poor electronic conduction in various traditional ceramic or even metal-based powders (e.g., aluminum powder, etc.). Due to its soft nature, carbon may serve as an efficient interphase material improving the actual contact area after exerting some external pressure during electrode preparation and/or multiplying the number of contacts.
The binder additive is in fact a mixture of several polymers and organic additives that combine critical multiple roles that are discussed throughout this article.
Tape casting is widely utilized in the electrode manufacturing processes. Basically, it contains the preparation through ball milling of a suspension (slurry) of AM, C and B in a volatile solvent. This slurry is then cast onto the current collector through a slot-die or a transfer-roll process. After drying, composite electrodes are usually pressed down to about 30–40% porosity using a rolling machine or hydraulic press. This treatment is essential for obtaining simultaneously high energy, high rate and good cycling stability. The composite obtained films are porous and electronically conducting. They are then impregnated by the liquid electrolyte during the battery assembly. For long-lasting battery operation, the composite electrode needs to be chemically and electrochemically stable. It also needs to maintain a good mechanical cohesion in the presence of the liquid electrolyte and during the volume changes which occur when inserting and extracting Li+ ions within the AM grains.
For optimum battery energy density, the amount per unit of mass and volume of the non-electroactive additives need to be minimized. Generally, C is chosen as an acetylene black or its mixture with graphite, and B is selected as either poly(vinylidene fluoride) (PVDF) or a mixture of carboxymethyl cellulose (CMC) and a rubber latex, and the AM:C:B mass ratio is generally close to 90:5:5.
While polymers have been thoroughly investigated for a while for their use as a component of the electrolyte, their utilization as the electrode binder has not been thoroughly studied, so that the literature is scarce on this subject. Many studies have been done probably in industry in this field because battery companies need to commercialize efficient electrodes, but such knowledge remains proprietary information and thus is not published. Results from Guy et al. are a striking example demonstrating that the electrochemical performance of the composite electrode may strongly depend on the selection of the polymeric binder [1]. As a matter of fact, a 50% increase of the cycling capacity of Li1.1V3O8 based electrodes was obtained after using instead of PVdF or PVdF-HFP a poly(ethylene oxide) (PEO) [1], or a poly(methyl methacrylate) (PMMA) [2], polymer that had been preplasticized by the solvent (ethylene carbonate, EC, + propylene carbonate, PC) of the liquid electrolyte or by the liquid electrolyte itself (EC+PC + lithium bistrifluorosulfonimide [LiTFSI]) during the fabrication step of the electrode, as indicated in Fig. 1. This study shows that designing an efficient electrode needs the tailoring of the binder.
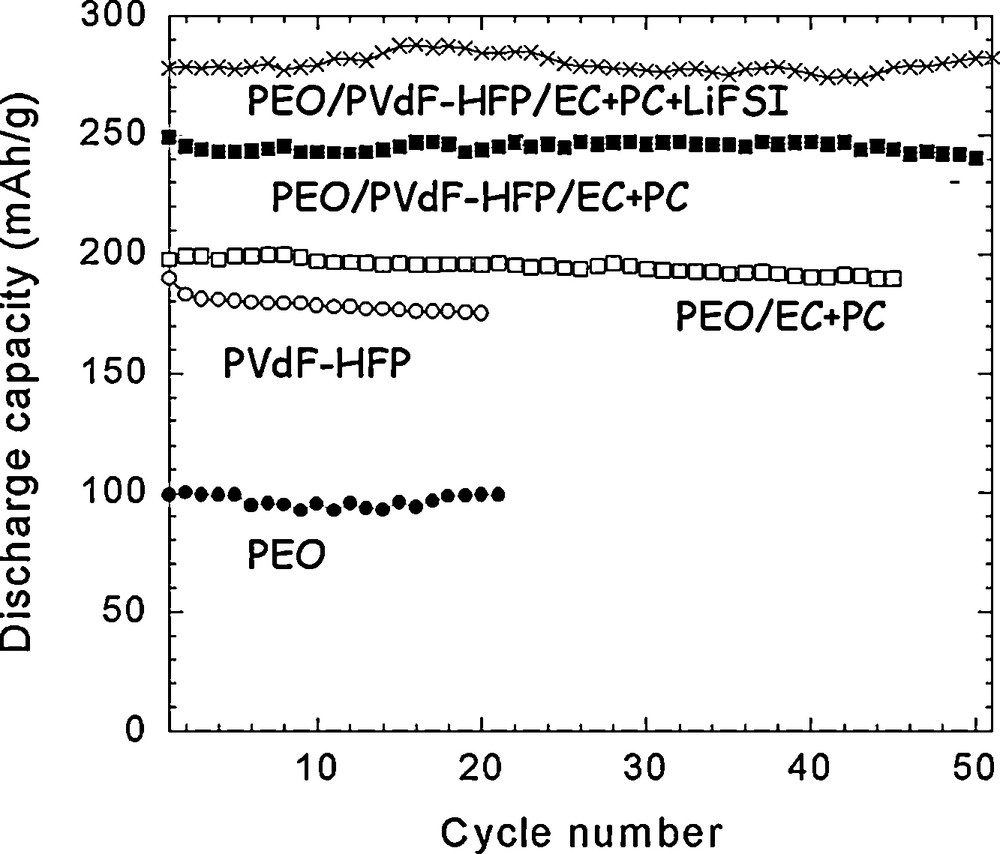
Discharge capacity (in mAh per g of active material) vs. cycle number at RT for different cells made with Li1.1V3O8-based positive electrodes prepared with different binders (using 1 M LiTFSI EC + PC electrolyte; 2–3.3 V; 20 °C; C/5 rate). This figure was edited from [1b], with permission from the Electrochemical society, Inc.
In the following, we will discuss the now well-established major roles that the polymeric binder plays: (i) in the dispersion process and stabilization of the AM and C powders in the electrode slurry during the composite electrode preparation, and thus on the architecture of the electrode; (ii) in the electronic and ionic conductivity of the composite electrode; and (iii) in the mechanical properties, i.e., the cohesion of the electrode and its adhesion with the current collector.
2 The usual and new binder combinations
Polymers have been mostly studied for their application as the electrolyte solvent of dry lithium (metal) polymer batteries [3–5] or as the separator (a microporous membrane) of LiB [6], rather than as the binder of composite electrodes. PEO is yet an exception because it has been used for more than 20 years as both the electrolyte host and electrode binder of dry lithium polymer batteries. But PEO is usually not utilized in composite electrodes for LiB because this polymer electrochemical stability window is limited to 4 V vs. Li/Li+, which is not suitable for high voltage positive electrodes such as LiCoO2. As a result, polymers with higher electrochemical stability such as poly(tetrafluoro ethylene), PTFE, or PVdF have been most widely adopted as the binder for composite electrodes in LiB [7–10]. A copolymer of vinylidene fluoride with hexafluoropropylene, PVdF-HFP, is used in both polymeric electrolyte and composite electrode of the plastic LiB technology [11].
A new trend is developing to substitute the PVdF binder. Some of the reasons that have been brought up include high cost, insufficient mechanical properties (strong binding strength, but low flexibility) and stability aspects. Moreover, recently, an attempt has been made to switch from the non-aqueous to the aqueous processing techniques, to reduce the cost, the safety and environment concerns associated with the use of the organic solvent of PVdF, i.e., N-methyl-2-pyrrolidone, NMP. Aqueous binders have thus gradually replaced PVdF for the negative electrode and are now examined for the positive electrode.
Examples of other binders than PVdF are PEO [1], PMMA [2,12], poly(acrylonitrile-methyl methacrylate) [13a], aromatic polyimides [14], and polypyrrole [15] for non-aqueous processing; gelatin [16], poly(acrylamide-co-diallyldimethylammonium chloride) (AMAC) [13b], and polyacrylic acid (PAA) [17] for aqueous processing. Combinations of several polymers have also been proposed for the aqueous processing: (i) CMC, styrene-butadiene rubber latex (SBR latex), and PAA [18]; (ii) acrylonitrile-butadiene (NBR) rubber latex and CMC [19]; (iii) polyacrylic rubber latex (LA132) and CMC [20], (iv) ammonium polyacrylic acid (PAA−NH4) and LA132 [21]; (v) NBR, CMC and the isooctylphenylether of polyoxyethylene [22]. In these combinations, the different roles of the binder, i.e., processing aid (thickener and dispersing agents), and adhesive are distributed over different polymers for higher performance. These new and efficient combinations were developed thanks to fundamental studies that clarified the relationships between the processing, the architecture and the electrochemical performance of the composite electrode.
3 Relationships between the processing, the architecture and the electrochemical performance of the composite electrode
The efficiency of the electrode architecture with respect to the transport of the charge carriers (electrons and lithium ions) toward the active matter is usually quantified by two macroscopic electrical properties, the ionic and the electronic conductivity. Particle size and intrinsic ionic and electronic conductivities are other important parameters with respect to transport inside active matter, which is not however, discussed in detail here. Ionic conductivity is a property difficult to measure for composite electrodes because it is masked by the electronic conductivity. The ionic conductivity κc is usually extrapolated from the bulk conductivity of the liquid electrolyte κo using
| (1) |
The electronic conductivity follows a percolation process in which the conductivity changes by 5–9 orders of magnitudes for a critical C volume fraction ΦC [1c,24]. The change in capacity also follows a percolation process in which a sharp increase in capacity is observed for the same critical C content. Thus, several studies aimed at predicting ΦC as a function of the shape and orientation of the C particles, film thickness [25], respective effective volume fractions of AM and C [26]. Guy et al. found significant variation in conductivity with the binder composition [1c], as this one was shown to have a strong impact of the dispersion state of the C particles within the electrode slurry and thereafter in the dry composite film (Fig. 2).
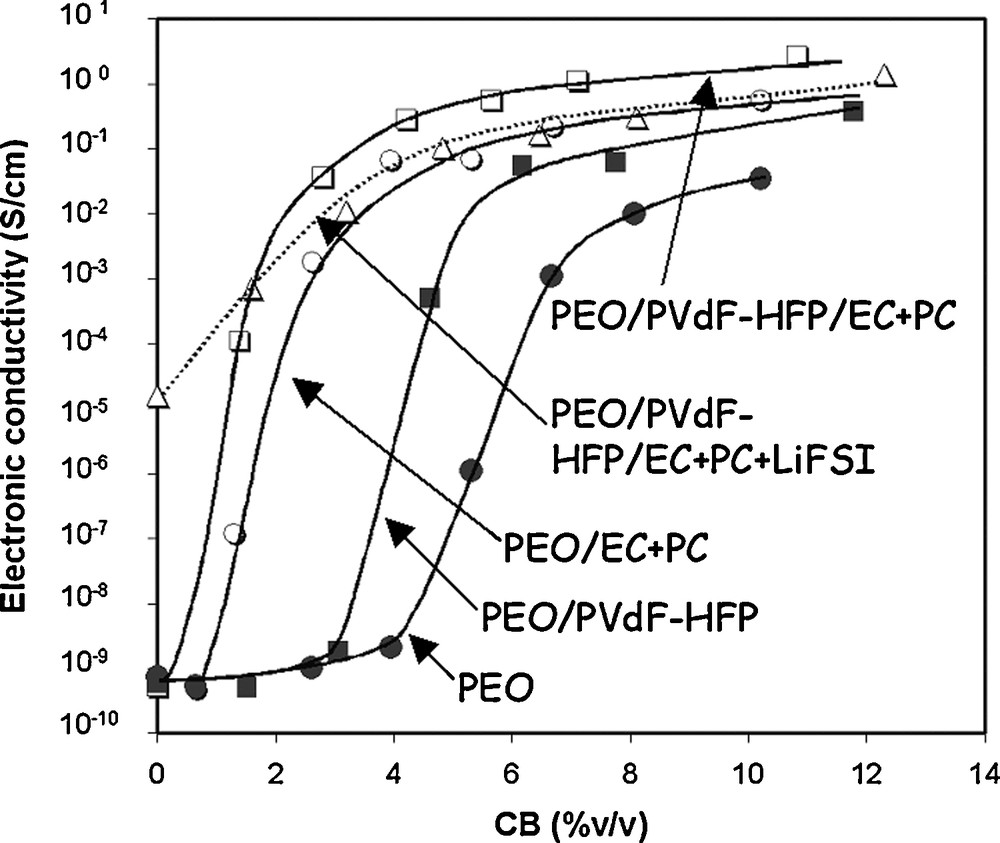
Electrical conductivity vs. CB composition for CB/binder blends with various binders. This figure was edited from [1c], with permission from the Electrochemical Society, Inc.
The study of the correlation between the electrochemical performance and the macroscopic transverse electronic conductivity (surface-to-surface average conductivity) shows that above ΦC, the electronic wiring of the AM grains, and as a result the electrochemical performance, are in fact limited by the number N of C/AM contact points at the AM grains surface [1c,2a,16,27], Fig. 3.
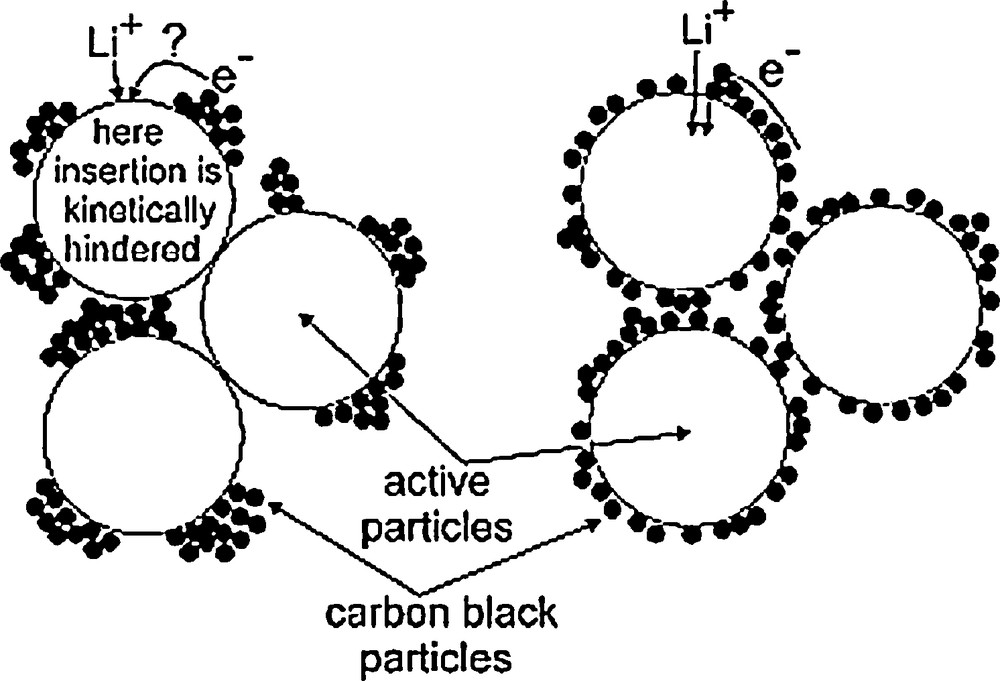
Schematic drawing indicating the impact of C distribution on the kinetics of Li+/electron electrochemical insertion into AM particles. This figure was reprinted from [16], with permission of Elsevier.
In addition to the electrode capacity, the homogeneity of the C distribution within the composite also affects the cycle life of the electrodes [28–31]. According to these authors, when the conducting additives are not uniformly dispersed, the AM grains are not evenly utilized and they also experience an unequal topological potential. Then some AM grains are degraded because they are overcharged–overdischarged, which leads to a gradual fade in capacity. The loss of contacts between the AM particles and the C network is another source of capacity fade [32]. This can occur when the binder is non-homogeneously distributed in the composite electrode [2c].
Because the agglomeration of either AM or C decreases the contact probability between them, it is essential to obtain a well-dispersed powder suspension during the electrode manufacturing process. The dispersion of electrode ingredients is controlled by several parameters such as mixing method [2c,33,34], material properties (such as AM and C particle size and shape, specific surface area and/or solvent uptake, density) [35], solid loading [2b], and interparticle interaction forces. These parameters affect the mixing efficiency in the slurry preparation step, the stability within the slurry upon casting and drying on the current collector and, consequently, the dispersion of the electrode components in the composite electrode layer. Polymers are essential to tailor interparticle interaction forces and achieve an homogeneous and structured architecture of the composite electrode.
4 Role of the binder on the processing
In preparation of the slurry, the solvent must wet the powders first. Mechanical work is then supplied to break down aggregates and agglomerates of particles. Finally, the particles must be stabilized against flocculation and settling. Colloidal powders of carbon black or nano-AM are difficult to disperse because they easily flocculate due to their large surface area.
It is convenient to distinguish three main types of interaction forces between the particles: (i) the van der Waals attraction is universal in all disperse systems; (ii) double layer interaction occurs between particles when a charge separation happens at the solid/liquid interface with the formation of an electrical double layer. This interaction is repulsive when the double layer is of the same sign in all particles. It becomes attractive if two types of particles with opposite double layer exist in the dispersion; and (iii) a third type of interaction (steric or osmotic) exists in the presence of adsorbed or dissolved polymer. Depending on the physicochemical properties of the suspending medium, polymer molecular microstructure and concentration, this interaction can be repulsive or attractive.
At rest, i.e., without any hydrodynamic shear stress applied, when the net force is attractive, flocculation occurs. On the contrary, stable dispersion can be formed when the net force is repulsive. For electrostatic stabilization, powders need to have a sufficient zeta potential to provide strong repulsion between particles. For steric stabilization, polymer adsorption is required to provide steric hindrance or osmotic repulsion between particles. However, an efficient stabilization requires dense polymer adsorbed layers, which are not suitable with good electrical contacts in the dried electrode, see Part 5 below.
In aqueous slurries, polyelectrolyte species, such as PAANH4 or CMC, which ionize in solution and attach on powder surfaces, combine the electrostatic and steric stabilization mechanisms with minimum amount added. The effect of such polyelectrolyte dispersants on the dispersion behavior of aqueous LiCoO2 slurries and their corresponding cell performances has been investigated by Li et al. [21]. The study of the rheological behavior of the electrode suspension showed the stabilizing effect of PAANH4. The strong decrease in viscosity shows that the interparticle interactions switches from attractive to repulsive when PAANH4 (6000 g/mol) is added (Fig. 4). Although increasing PAANH4 content could increase the dispersion homogeneity of powders, too much addition of PAANH4 led to a negative effect on the adhesion strength, electronic conduction, and consequently on electrochemical performance. It can be seen in Fig. 4 that the threshold amount for enhancing cell performance is in a range from 0.01 to 0.02 wt.%. The electrode contained 92.3 wt.% LiCoO2 (μm-size), 6.1 wt.% graphite (μm-size), and 1.6 wt.% LA132 binder.
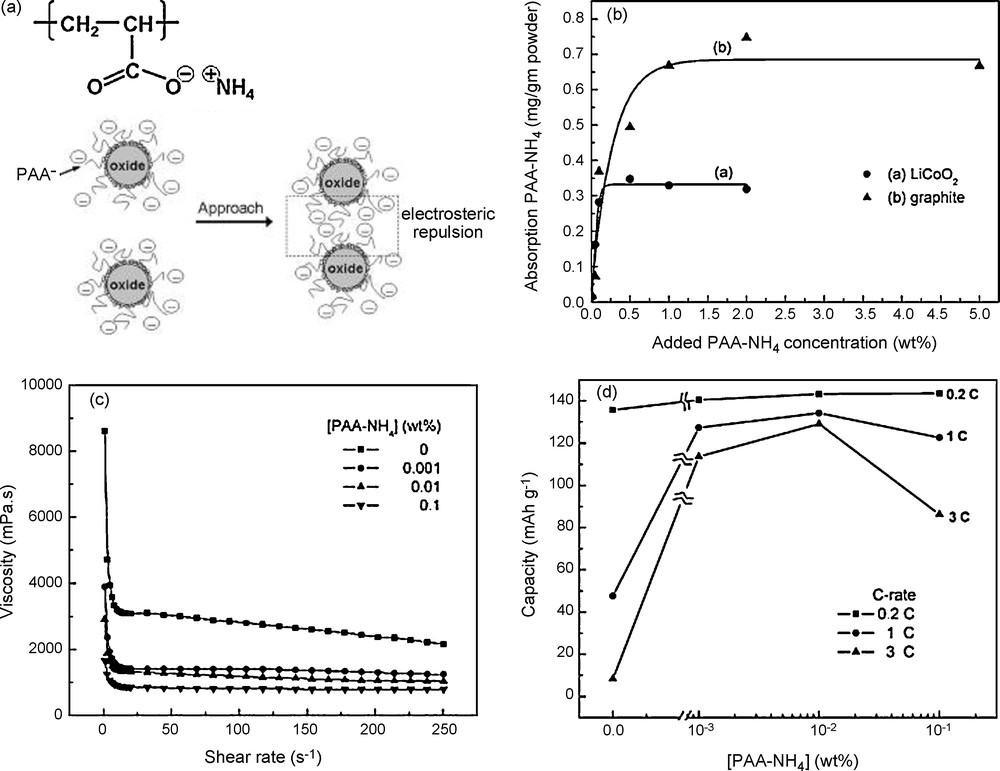
(a) Drawing showing the chemical structure and stabilization mechanism of PAANH4. (b) Adsorptions of PAANH4 on LiCoO2 and conductive particles (c) Viscosity as a function of shear rate for 68 wt.% aqueous LiCoO2 slurries with PAANH4 concentrations of 0–0.1 wt.%. (d) Discharge capacity at various discharge rates for LiCoO2/Li half-cells as a function of PAANH4 concentration. This figure was edited from [21], with permission from the Electrochemical society, Inc.
CMC (330,000 g/mol) has been shown to work well as a dispersing agent for the LiFePO4 (average particle diameter of 300–500 nm) [36] and Natural Graphite (5–20 μm) [18]. The adsorption of CMC contributes to charge development at the surface of the AM particles through the same mechanism as depicted in Fig. 4a as a result of the presence of the carboxylate functional groups within the adsorbed layer of CMC. A lower degree of substitution (DS) on CMC improved the quality of the LiFePO4 and Graphite dispersions and its maintenance over months, as well as a longer cycle life, presumably due to enhanced interactions with AM particles and adsorption onto AM particles. In these studies, the CMC content in the electrodes was 4 and 1.5 wt.%, respectively.
CMC is also used as a thickener. A thickener is added to give to the electrode slurry proper flow properties with respect to tape casting. Ideal flow behavior is: (i) a low viscosity at high shear-rate to allow a good mixing during the dispersion step; and (ii) a very high viscosity when the slurry is left to rest in order to prevent settling during drying. Carbohydrate type polymers such as cellulose derivatives are particularly suitable as thickeners. Indeed, their rather inflexible cellulosic backbone, compared, for example, with vinyls and acrylics, causes these polymers to be in outstretched form in solution, which, in turn, results in rather high viscosity levels at low shear-rate. However, the facile alignment of the chains under flow results in marked shear-thinning properties, i.e., a strong decrease of viscosity with increasing shear-rate.
Porcher et al. compared CMC (250,000 g/mol) and Hydroxypropylmethyl cellulose (86,000 g/mol) as thickening agents [22a]. After drying, an homogeneous morphology is achieved in a CMC-based electrode, contrarily to the HPMC-one, where a concentration gradient is observed, Fig. 5a–b. Poorer rate capability is obtained with HPMC, while the CMC-based electrode shows identical performance to the standard PVdF-based electrode. The study of the rheological properties demonstrates that when the thickener is CMC, the electrode slurry displays a solid-like behavior soon after having been tape cast, likely due to the build-up of a network of the colloidal CB particles, spanning the whole system and bridged by CMC chains, Fig. 5c–d. Contrarily, when the thickener is HPMC, the electrode displays a liquid-like behavior and the settling of the heaviest particles (LiFePO4) results in a concentration gradient and poor electronic wiring. The ionization of the carboxylate groups of CMC appears to be favorable to an optimal adsorption and conformation of the polymer with respect to the bridging mechanism.

Scanning electron microscopy (SEM) pictures of the cross-section of (a) the HPMC and (b) the CMC electrodes. Schematic interpretation of the interactions and dispersion states within the (c) HPMC and (d) CMC electrode slurries. This figure was edited from [22], with permission from the Electrochemical Society, Inc.
The study of Porcher et al. shows that knowledge of the physical chemistry of polymers in solution can be used to control the interactions of the particles within the slurry and tailor the electrode architecture. A very elegant utilization of this concept has been proposed by Gaberscek et al. [16]. In their work, the AM particles are first pretreated in a gelatin solution. Through careful control of the composition of the gelatin solution and experimental conditions (temperature, pH, ionic strength, etc.), tailored amounts of gelatin can be adsorbed to the surface of the AM particles. The adsorbed gelatin can then capture the C particles so that AM are uniformly covered by C particles (Fig. 6).
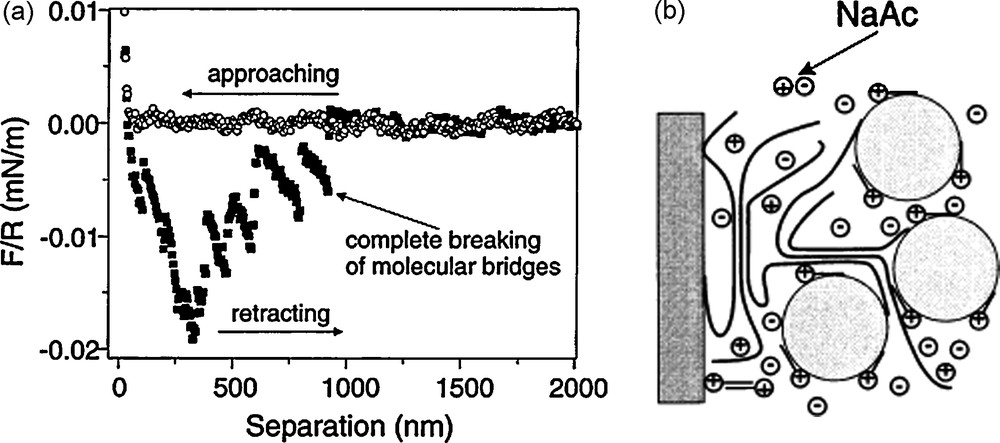
(a) Force between glass sphere coated with gelatin and carbon black coated glass surface in 1 mM cetyltrimethylammonium bromide (CTAB) and 0.05 M sodium acetate (NaAc) water solution. (b) Schematics of the capturing of carbon black particles on gelatin-modified surface. This figure was edited from [37], with permission from Elsevier.
In non-aqueous slurries, the same principles hold. The magnitude of the electrostatic forces is, however, extremely sensitive to the permittivity of the suspending medium and the osmotic/steric forces depend on the affinity between the polymer chain and the solvent. Nevertheless, the tailoring of the interactions between all the constituents, i.e., AM, C and B, during the electrode processing can be achieved by tuning the solvent properties [2b–d,38].
5 Role of the binder on the electronic conductivity
Above ΦC, i.e., for C content of usual composite electrodes, Guy et al. [1c] found that the conductivity follows a unique relationship, independent of the binder combination and of the AM presence, Fig. 7
| (2) |
| (3) |
| (4) |
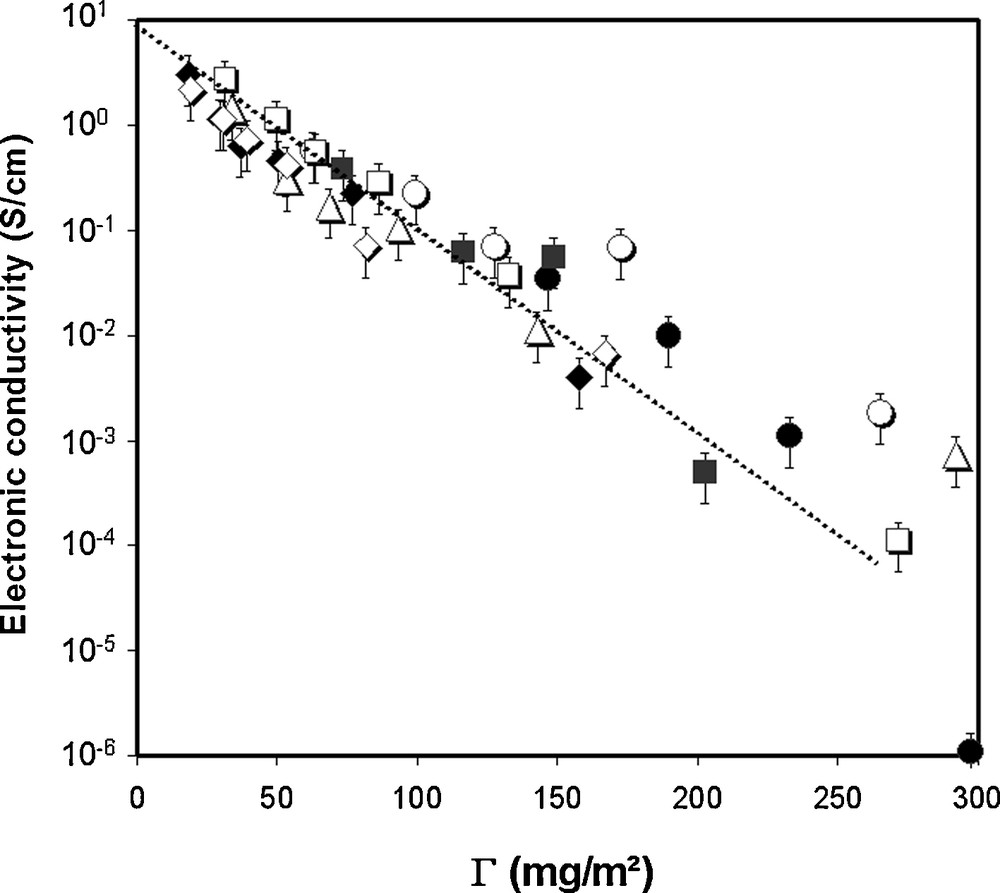
Electronic conductivity vs. Γ (ratio of mass of polymer to C total surface area as estimated through BET surface measurements) for different composite electrodes. This figure was edited from [1c], with permission from the Electrochemical society, Inc.
The experimental law for electronic conductivity found by Guy et al. (Eq. (3)) shows similar dependence of the electrode electronic conductivity on ΦBinder/ΦC. Thus, this similarity does support that the electronic transport in composite electrodes is due to a tunneling mechanism through the insulating polymer layers in between the conducting particles. Noteworthy, in the study of Guy et al., PEO/PVdF-HFP binders mixture were studied [1c]. It was observed that PVdF-HFP, which did not adsorb to the surface of the particles, because it was not soluble in the processing solvent, played no role on the electronic conductivity of the composites. The dependence logσ ∝ ΦPEO/ΦC was observed whatever the PVdF-HFP composition in the PEO/PVdF-HFP blend. Thus, in Eq. (4), the term binder holds for the one of the polymer(s) of the binder combination that can adsorb to the surface of the C particles.
Because the binder is likely to adsorb on the AM particles the same way as on C, it is likely that the resistivity of the C/AM electrical contacts depends on the same parameters, i.e., thickness of the adsorbed polymer layer. Although Fig. 7 suggests a minimum amount of binder is better for electronic conductivity, it has been observed however that a lack of binder may result in poor dispersion and/or insufficient of mechanical stability upon cycling of the AM/C contacts and of the electrode/current collector interface [2b,c,45].
6 Role of the binder on the ionic conductivity
According to Choi et al. [12], the amount of liquid electrolyte uptake into the electrode and the electrode ionic conductivity increase with the binder content and with the binder/electrolyte affinity – that is a key factor that play on the wettability of the composite electrode by the liquid electrolyte. According to Sugita et al. [46] and the approach based on the thermodynamic work of adhesion, polar binders such as PVdF, PMMA and CMC improve the composite electrode wettability compared to neutral binders such as SBR or polyethylene (PE). As a matter of fact, a higher Li-ion conductivity has been measured for the polar 2-ethylhexyl acrylate-acrylonitrile (2EHA-AN) copolymer compared to the neutral SBR, both swollen by electrolyte solution [47]. As a result, the LiCoO2 electrode prepared with 2EHA-AN copolymer showed highest capacity, suggesting that Li+-ion conduction inside the elastomeric binder contributes to the enhancement of electrochemical performance. In addition, a recent work from Sisbandini et al. [48] suggests that the coating of AM particles with short-chain polymers (polyetheramine [PEI]) could mediate the polarity difference between the AM particles and the electrolyte, thereby enhancing Li+ charge transfer, Fig. 8.
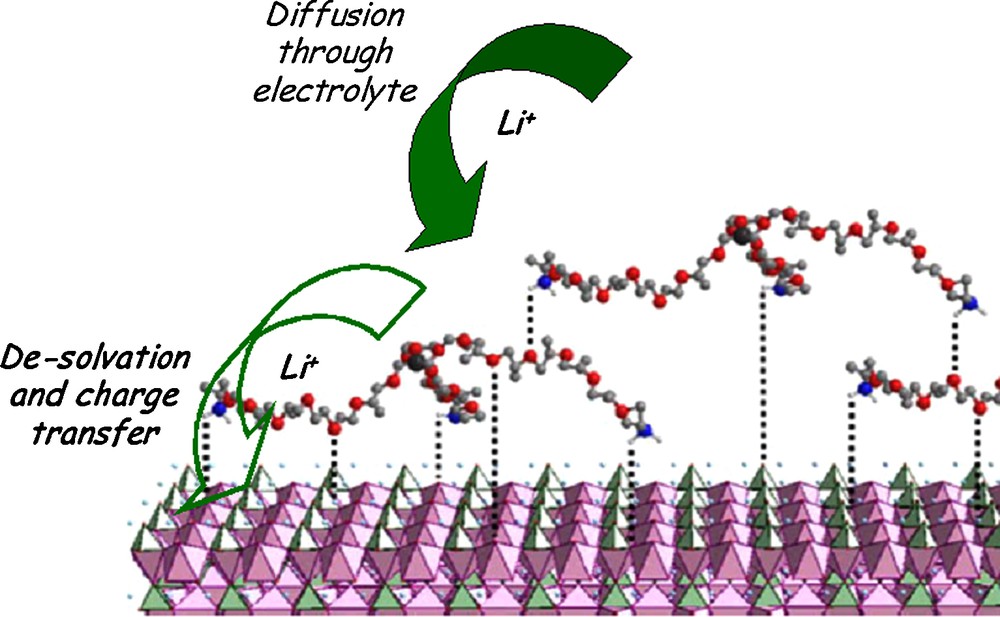
Schematics of the mediation of the electrolyte/electrode interaction across the interface region by polyetheramine. Dotted lines shows interactions postulated between ether O of PEI with positive sites on the LiFePO4 surface, Li-O or Fe-O coordinations. This figure was edited from [42], with permission from the Electrochemical society, Inc.
7 Role of the binder on the mechanical properties
The understanding of the “binding strength” of the binder remained qualitative for a long time although it is its original function. In most papers, binding refers to the binder property of giving to the electrode a good mechanical strength (cohesion) and a good adherence to the current collector. These two mechanical properties are usually measured using the well-known tensile and peel tests, which give the macroscopic maximum elongation and stress (tensile strength) at break and the debonding force (peel strength). Babinec et al. studied the failure mechanism of PVdF- and LiCoO2-based positive electrodes [8]. They showed that the tensile strain at break is less than 2% and that particle delamination from the PVdF matrix is the mechanism for stress relief. This critical 2% of deformation is likely to be reached in the battery assembling operations.
Yoo et al. extensively studied the relationship of the PVdF binder interaction with graphite, the processing conditions, and the binder morphology within the negative composite electrode and the mechanical strength of the composite electrode [10]. Graphitic and amorphous carbons with different particle size and surface area and PVdF unmodified or modified with hydroxyl (−OH) groups were studied. The interaction was shown to depend on the carbon particle surface chemistry and on the chemical structure of PVdF. The binder mainly formed weak hydrogen bonds between its fluorine atoms and hydrogen atoms from graphite. A stronger bond of intermediate binding energy between semi-ionic and covalent C−F occurred between PVdF and amorphous carbon. Due to all these interactions, PVdF was found to adsorb to the surfaces of the carbonaceous particles. The amount and the morphology of the adsorbed PVdF depended on the interactions. The polymer did not cover the whole surface of carbon particles, but formed discrete polymer domains. The degree of homogeneity of PVdF distribution on the composite film could be attributed to the particle surface chemistry. The large particles having small surface areas, and therefore relatively few interacting sites with PVdF, generated a weak interaction between graphite and the polymer, leading to aggregation of PVdF instead of more homogeneous distribution. The −OH functionalized PVdF showed higher amount of absorption with carbon particles through enhanced hydrogen bonding capability. The mechanical strength of the studied negative electrodes bonded to the copper current collector was shown to depend on the structure of the carbon particles and on the chemical properties of the PVdF binder. Amorphous carbon, which forms a strong C−F bond, shows much higher mechanical strength than crystalline carbon. This study is in agreement with the work of Despotopoulou et al. which shows the adhesion of graphite negative electrodes onto the copper current collector can be strongly improved by using functionalized PVdF with maleic anhydride [7].
Porcher et al. [22b] observed a correlation between the maximum calendaring pressure before debonding from the current collector of a composite electrode as a function of the ratio between the amount of binder in the electrode and the apparent surface of the grains (as measured through laser granulometry), Γ*, Fig. 9. The electrode contained LiFePO4, carbon black, CMC, NBR and the isooctylphenylether of polyoxyethylene.
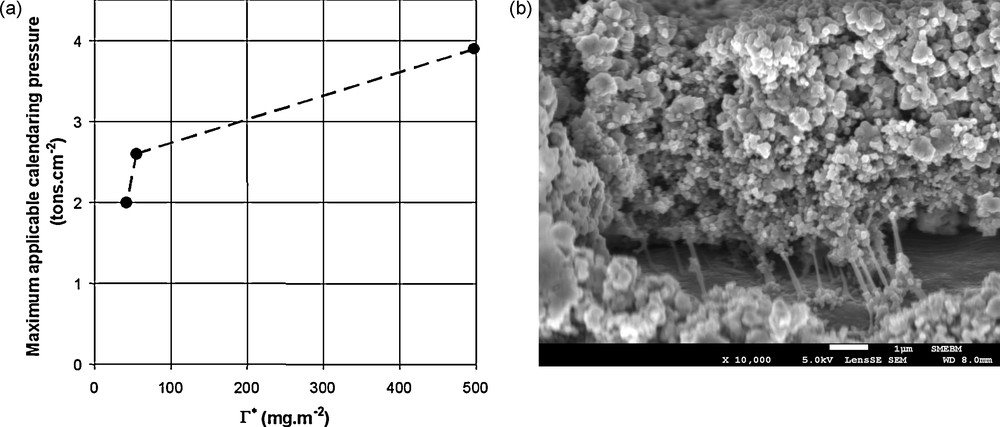
(a) Maximum calendaring pressure before debonding from the current collector of a composite electrode as a function of the ratio between the amount of binder in the electrode and the apparent surface of the grains (as measured through laser granulometry). (b) SEM picture of a damaged interface between an electrode and a current collector.
All these results can be rationalized with recent model developed for fracture of polymer interfaces [49]. Mechanical toughness in polymer-based composite materials has been shown to be controlled by the strength of the polymer bridges at interfaces
| (5) |
8 Role of the binder on the SEI layer
When graphite powders are utilized as the AM, the irreversible capacity appears at the first cycle, because reductive decomposition of the electrolyte solution occurs at the graphite/electrolyte interface during the first charge (electroreduction) including the formation of solid electrolyte interphase (SEI). The existence of an SEI layer plays an important role as the kinetics of lithium intercalation is predominantly determined by the SEI, because all lithium ions in the electrolyte solution must cross the interphase accompanied with desolvation. A few studies report on the strong influence of the binder chemistry on the surface film formation on carbonaceous electrodes. Zhang et al. report that the AMAC aqueous binder assists in forming a more conductive SEI layer than PVdF [13b]. Drofenik et al. and Buqa et al. observed that graphite electrodes with CMC binder showed less irreversible capacity in the first cycle, as compared with that using PVdF binder [50,51]. In another study, El Ouatani et al. showed CMC favorably participates to the SEI on Graphite electrodes, likely due to the chemical reactivity of its hydroxyl groups −OH towards the electrolyte [52]. Komaba et al. showed that compared to PVdF, the efficiency at the initial cycle was improved by PAAH and alkali polyacrylates (PAALi and PAANa) [53]. Graphite powders were observed to be more uniformly covered with a polyacrylate thin layer, unlike PVdF, resulting in a lower area of direct contact between the graphite and the electrolyte solution. The authors also suggested that the carboxyl groups C(O)OH or C(O)O− of the polyacrylates incorporated in the SEI layer enhance the desolvation of the Li+ ions to reduce the initial irreversibility. The alkali polyacrylates led to higher efficiency than PAAH due to a more aggregated conformation of PAAH through hydrogen bonding.
9 Which binder for the future negative electrodes?
Commercial LiB based on the graphite/LiCoO2 chemistry are approaching the theoretical limit for volumetric energy density. The ever-growing demand for LiB of higher energy density, has lead to extensive research into materials of higher capacity. Silicon is an attractive alternative to graphite due to its high theoretical capacity of 3579 mAh per gram of Si, which corresponds to the reaction 15Li+ + 4Si + 15e− → Li15Si4 [54]. However, a large capacity fade is observed during cycling, as the result of large volume change in the material, during alloying and dealloying with lithium. Degradation of Si composite electrodes is a direct consequence of the breakdown of contacts between Si and C particles [55,56]. The choice of binder has been shown to be very critical to the cycling performance of silicon and other alloy negative electrodes [45,57–62] Chen et al. suggested that cycling stability might benefit from increasing the deformability of the binder material [57]. They successfully synthesized a poly(vinylidene fluoride-tetrafluoroethylene-propylene)-based elastomeric binder system combined with an adhesion promoter that improved the cyclability of amorphous Si–Sn alloy. Buqa et al. and Liu et al. reported that the cycle life of Si-based electrodes was improved by using an aqueous binder containing the elastomeric styrene butadiene rubber (SBR) and sodium CMC [51,58]. However, Li et al. showed that the use of the stiff CMC binder resulted in better capacity retention than the SBR + CMC combination [59]. Lestriez et al. showed that the efficiency of the CMC could be attributed to a more efficient bridging of Si and CB particles during the electrode elaboration, coming from the CMC extended conformation in solution combined with its adsorption on both Si and CB particles [60a]. And Mazouzi et al. and Hochgatterer et al. showed that CMC was able to chemically bond with a Si surface through a condensation mechanism between the carboxylic acid C(O)OH groups of the CMC and the silanol SiOH groups present at the surface of silicon during the drying step of the electrode preparation [60b,61].
All these results can be interpreted by considering Eq. (5), as the bridging efficiency of CMC increases Σ and the chemical anchoring of CMC onto the silicon particles increases fb, both parameters contributing to increase the mechanical stability of the Si/C contacts. However, a recent study by Key et al. [63] suggests the grafting of CMC stabilizes the surface of silicon particles with respect to some unfavorable irreversible electrolyte reductions that might be the main reason for better cycling performance of CMC-based silicon electrodes.
10 Conclusion
The new binders that are replacing the standard PVdF are combinations of polymers to satisfy the several roles played by the binder: (i) in the dispersion process, where it acts both as dispersing agent and as a thickener; (ii) in the mechanical properties (cohesion and adhesion) that depend on the area density of adsorbed/grafted chains that bridge the particles altogether and with the current collector; (iii) in the electronic conductivity of the electrode, as the electrons have to tunnel through or near to the polymer chains that maintain the contacts upon cycling; (iv) in the ionic conductivity of the electrode, as it modifies the wettability of the powders and the desolvation process of the Li+ ions at the AM particle surface/electrolyte interface; (v) in the chemical and electrochemical stability of the same interface. All these properties critically depend on the very thin layer formed by the polymer chains at the surface of the AM and conducting agent particles. That is where research efforts have to focus on, now, for better understanding and further optimization of electrode performance through tailoring of the binder combination.
Acknowledgements
The author gratefully thanks his coworkers, D. Guyomard, J. Gaubicher, P. Moreau, N. Dupré, J.-C. Badot, R. Bouchet, M. Deschamps, J.-L. Monfort, A. Nazri, D. Plée, S. Levasseur, D. Guy, E. Ligneel, W. Porcher, C. Fongy, K. Seid, V. Gaudefroy, P. Mongondry, S. Desaever, D. Mazouzi for friendly and fruitful discussions.