

1 Introduction
Oxide ion conducting materials, in which oxygen can easily migrate at high temperature, are the heart of solid oxide fuel cells (SOFCs) and high temperature electrolysers (HTE). For these applications, the electrolyte consists of a dense ceramic with electrodes on both faces. Whereas pure oxide conduction is required for the electrolyte (to avoid any short circuit), mixed conductors (ionic and electronic) are usually preferred for the electrode materials. Because of its high stability, yttrium stabilised zirconia (YSZ) remains the most developed electrolyte material, but it suffers from a too low conductivity at temperature lower than 700 °C. The electrolyte ceramic must fulfil several requirements. It must be dense, it has to be a pure oxide ion conductor and stable under oxidative and reductive conditions.
In this study, with the aim to evidence new oxide ion conductors, in a first step, the less reducible elements were identified, then, the phase diagrams corresponding to these elements were analysed, known structures were listed, most favorable structures for oxygen diffusion (displaying chains of polyhedra connected with oxygen atoms, for instance) were selected, and, finally, the possibility of defect chemistry (partial substitution of an element by another with the aim to introduce oxygen vacancy or interstitial in the structure) was considered. This led us to consider the La2O3-Ta2O5 system and this article will focus on the La3TaO7 structure, which is of Weberite type. In a first approach, atomistic calculations were carried out with the aim to identify the possibility of defect chemistry and the most promising substituting cations. In a second step, a solid solution La3−xSrxTaO7−x/2 was confirmed by X-ray diffraction and Raman spectroscopy and the impact of partial substitution for La with Sr on conduction properties was investigated by impedance spectroscopy.
2 Experimental
La3TaO7 and La3−xSrxTaO7−x/2 (x = 0, 0.05, 0.10, 0.15, 0.20, 0.25, 0.30) compounds were prepared by solid state reaction at 1600 °C for 140 hours with intermediate grindings in alumina crucibles from the mixture of the corresponding oxides and carbonate weighted in stoichiometric amount: La2O3 (Sigma Aldrich – ≥ 99.9%), Ta2O5 (Sigma Aldrich – ≥ 99.9%), SrCO3 (Sigma Aldrich – > 98%). At such high temperatures, the risk of reaction with the alumina crucible could not be neglected. Indeed, in the case of preparation of a large amount of the sample (40 g), which necessitates additional annealing to obtain pure phase, a small reaction was noticed. Refinement of neutron diffraction data (not presented here) revealed LaAlO3 as impurity, but its content was less than 1%. However, no trace of aluminium was evidenced by X-ray fluorescence when small amounts of the sample (3 g) were prepared.
The purity of the as prepared compounds was characterised by X-ray diffraction using a Hüber diffractometer equipped with an image plate-sensitive detector over the 10–100° 2θ domain with a counting time of 15 minutes (CuKα1 = 1.54056 Å).
Raman spectra were recorded at room temperature using the 647.1 nm excitation line from a spectra physics krypton ion laser. The beam was focussed onto the sample using the macroscopic configuration of the apparatus. The scattered light was analysed with an XY Raman Dilor spectrometer equipped with an optical multi-channel charge-coupled device liquid nitrogen-cooled detector. Acquisition and data processing were performed with the LABSPEC software from the Horiba Jobin Yvon Society.
Samples were densified by Spark Plasma Sintering (SPS) at the “Plate-forme nationale de Frittage Flash PNF2”, in Toulouse, France. Papyex® was used to protect the sample from the die walls; it was removed by annealing at 1100 °C for ten hours. It is worth noting that before annealing, the pellet was dark inside (likely graphite pollution) but the initial colour was recovered after annealing at 1100 °C.
Impedance spectroscopy was performed in Warsaw, Poland, using a Solartron 1260 Impedance/Gain Phase Analyser associated to a Keithley 428 Current Amplifier in the frequency range 0.01 Hz 10 MHz (between 18 and eight frequency points per decade), with an applied AC signal of 30 mV rms. Platinum electrodes were sputtered by cathodic discharge on the two pellet faces. Impedance spectra were collected in air over two cycles of heating and cooling from 100 to 700 °C, every 20 °C, at stabilized temperatures (20 minutes stabilization time before each measurement). To verify the stability of the sample response, impedance spectra were measured repeatedly with a 1% drift tolerance and five successive measurements at maximum.
3 Results and discussion
3.1 The La3TaO7 structure in the La2O3-Ta2O5 phase diagram
The La2O3-Ta2O5 phase diagram consists of four defined compounds, La3TaO7, LaTaO4, LaTa3O9 and La2Ta12O33 [1]. Among these compounds, the structure of La3TaO7 is built upon chains of Ta-O octahedra connected by one oxygen atom in between chains of La-O and isolated lanthanum atoms (Fig. 1). It was described in the Cmcm space group [2,3]. There are three oxygen sites in the structure, O(3) sites belongs to the La(O) chains, whereas O(1) and O(2) sites correspond to the equatorial and apical site of the Ta(O)6 octahedron, respectively. When just taking into consideration the cationic lattice, this structure could also be viewed as an oxygen defective fluorite with one oxygen vacancy per La3TaO7 formula. However, because tantalum prefers a six-fold coordination, oxide positions in the structure are rather far from the ideal tetrahedral sites of the fluorite, and a simple calculation with the software “atoms” [4] led us to evidence two cavities located at (0.229, 0, 0.5)Ci1 and (0, 0.1795, 0.7436)Ci2 (Fig. 1). However, these two sites, with distances to the other oxygen sites lower than 2.3 Å (dCi1−O(2) = 2.21 Å, dCi1−O(3) = 2.06 Å, dCi2−O(2) = 2.29 Å taking [3] as structural model) are too small to accommodate an additional oxide ion. Nevertheless, one could expect that partial substitution of lanthanum or tantalum atoms could expand the cell. Moreover, a judicious choice of substitution could lead to the creation of defects in the structure, especially oxygen vacancies, which could increase the oxide ions mobility and, so, the conducting properties. With the aim to define the most relevant dopants, atomistic simulation was performed using the General Utility Program (GULP) code [5].
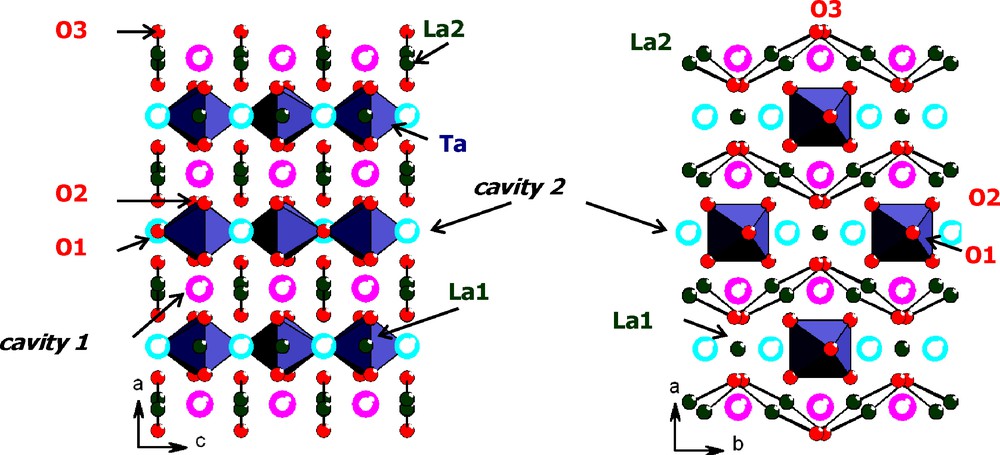
La3TaO7 crystalline structure in Cmcm space group with evidence of two cavities.
3.2 Defect chemistry in the La3TaO7 structure: atomistic simulation
The GULP code is a way to calculate various parameters characteristic of structures (atomic positions, dielectric and elastic constants) and to determine relevant defect energies. It is based upon energy minimisation using the Newton Raphton method and the Mott-Littleton methodology. An important feature of these calculations is the treatment of lattice relaxation about the point defect or dopant ion. In the Mott-Littleton approach, the crystal is divided into two regions, so that ions in a spherical inner region surrounding the defect are relaxed explicitly, and the remainder of the structure is treated by approximations [6,7]. The interatomic interactions are divided into long range (Coulomb) and short range (Buckingham) forces. The interatomic potential is given by:
| (1) |
| (2) |
The introduction of charged defects in the structure implies the polarisation of the ions, which is associated with a deformation of the polarisable ions. To take into account the ionic polarisability, the shell model developed by Dick and Overhauser, is used [8]. Each ion is considered as a core (charge X) connected by a string (constant k) to a shell (charge Y, no mass). The charges X and Y are chosen, so that their sum is equal to the ion charge.
For calculations, the Aij, ρij, Cij parameters (Eq. (2)) have to be determined for each short distance interaction between the two considered ions and the k, Y parameters, for each ion.
The interatomic potentials and the shell model parameters considered for La3TaO7 are given in Table 1. They were extracted from Ruiz-Trejo et al. and Khan et al. [9,10] for La-O and O-O interactions, and Donnerberg et al. [11] for Ta-O. For O-O interactions, potentials given by Fisher and Islam [12] and Pirovano et al. [13] were also tested; they led to similar conclusions.
Interatomic potentials used for La3TaO7.
| Interaction | A (eV) | ρ (Å) | C (eV.Å6) | Y(e) | k (eV.Å−2) |
| La3+…O2− | 1545.21 | 0.359 | 0 | −0.25 | 145 [9,10] |
| Ta5+…O2− | 1315.57 | 0.36905 | 0 | −4.596 | 5916.77 [11] |
| O2−…O2− | 22764.3 | 0.149 | 43 | −2.239 | 42 [9,10] |
In a first step, the Weberite structure was reproduced. The two models proposed by Rooksby et al. [2], on one hand, and Wakeshima et al. [3], on the other hand, converged to the same solution. The simulation led to a lattice energy Elattice = − 1416.4 eV/unit cell.
The relaxed unit-cell parameters and atomic coordinates are compared to the initial structural models in Table 2. They are in rather good agreement with the experimental data, excepted for O(1) coordinate which is shifted towards y = 1/2 which leads to less distorted Ta-O octahedral chains than in reality. Attempts at calculations with other O2−-O2− potentials [12,13] led to the same feature, only a difference in the values of unit-cell parameters was observed. Since the aim of the atomistic study here was just to define the possibility of defects as a preliminary study to direct the experimental work, no further optimisation of the structure was carried out. Since distortion of Ta-O octahedra is likely to be a key point in the oxygen diffusion mechanism, being aware of the limitation of our model, we did not attempt to calculate energy associated to oxygen migration, except Frenkel energy.
Unit-cell parameters and atomic positions of La3TaO7 in the Cmcm space group: experimental [2,3] and calculated data.
| Data from 1964 [2] | Data from 2004 [3] | Calculated | |||||||
| a (Å) | 10.8600 | 11.1863(4) | 11.123(5) | ||||||
| b (Å) | 7.7000 | 7.6152(3) | 7.904(7) | ||||||
| c (Å) | 7.8400 | 7.7556(3) | 7.667(8) | ||||||
| x | y | z | x | y | z | x | y | Z | |
| La(1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| La(2) | 0.22900 | 0.29400 | 0.25 | 0.2261(1) | 0.2992(2) | 0.25 | 0.25(1) | 0.25(1) | 0.25 |
| Ta(1) | 0 | 0.5 | 0 | 0 | 0.5 | 0 | 0 | 0.5 | 0 |
| O(1) | 0 | 0.43400 | 0.25 | 0 | 0.439(3) | 0.25 | 0 | 0.50(1) | 0.25 |
| O(2) | 0.12300 | 0.31600 | –0.03300 | 0.118(1) | 0.321(1) | –0.045(1) | 0.12(3) | 0.32(2) | 0.00(1) |
| O(3) | 0.13600 | 0.02400 | 0.25 | 0.169(1) | 0.027(2) | 0.25 | 0.135(1) | 1.00(1) | 0.25 |
Then, in a second step, defects were introduced in the structure. Energy associated to the formation of oxygen vacancies (lattice energy of the structure without an oxygen atom to simulate the extraction of an oxygen atom to infinity) on the three possible sites of the structure was first considered. Calculations led to oxygen vacancy energies of 19.39 eV, 19.45 eV and 19.16 eV for O(1), O(2) and O(3) sites, respectively.
The possibility to introduce an interstitial in the cavities of the structure was also taken into account. It led to negative values of (O″ci1) = −12.84 eV and (O″ci2) = −11.66 eV for both sites.
From these data, the Frenkel energy (energy associated to the migration of an oxide ion to the interstitial site, leading to the formation of an oxygen vacancy) was calculated:
| (3) |
A rather high energy of 7 eV was obtained. It was too high to imagine that a migration mechanism of an oxide ion from a crystallographic site to an interstitial vacant site would be possible. This was later confirmed by the low conductivity of La3TaO7 composition.
Therefore, with the aim of increasing the oxygen conductivity, partial substitution for cation in the structure was considered to increase the number of either vacancy, or interstitial site in the structure. Although substitutions with isovalent cation should not introduce any additional defect in the structure, calculations were also carried out with such cations. Based on the Kröger-Vink notation, the following reactions of substitution were considered, being an interstitial oxygen atom and an oxygen vacancy.
Substitution on the lanthanum sites (La(1) and La(2)):
| (4) |
| (5) |
| (6) |
| (7) |
| (8) |
Substitution on the tantalum Ta site:
| (9) |
| (10) |
| (11) |
| (12) |
| (13) |
Depending on the dopant oxidation state and on the type of substituted site, defects associated to charge compensation (vacancies, oxygen atom interstitials) are introduced. The energy of these reactions, called “solution energy”, is calculated by combining the lattice energy of the considered oxides and the associated defect energy.
The calculation was performed on a wide range of dopants, even with reducible ones, which were not considered for the rest of the study. The lattice energy corresponding to the corresponding oxides and interatomic potentials for each dopant are given in Tables 3 and 4.
Lattice energy of oxides corresponding to the dopants studied in this work.
| Oxides | Lattice energy Ul (eV) |
| La2O3 | −129.06 [10] |
| Ta2O5 | −317.51 [11] |
| CaO | −35.95 [14] |
| MgO | −41.29 [14] |
| NiO | −41.58 [14] |
| PbO | −34.69 [13] |
| SrO | −33.42 [14] |
| CuO | −43.79 [15] |
| Nd2O3 | −129.22 [16] |
| Bi2O3 | −139.39 [13] |
| Y2O3 | −134.74 [16] |
| Yb2O3 | −136.76 [16] |
| Sc2O3 | −144.47 [16] |
| Al2O3 | −160.5 [14] |
| TiO2 | −112.45 [17] |
| ZrO2 | −109.76 [18,19] |
| CeO2 | −105.66 [18] |
| SnO2 | −110.68 [16] |
| Nb2O5 | −322 [20] |
| MoO3/WO3 | −212.67 [13,21] |
Interatomic potentials used for the dopants studied in this work.
| Interaction | A (eV) | ρ (A°) | C (eV.Å6) | Y (e) | k (eV.Å−2) |
| Ca2+…O2− [14] | 1228.9 | 0.3372 | 0 | 1.26 | 34 |
| Mg2+…O2− [14] | 821.6 | 0.3242 | 0 | 2 | 99999 |
| Ni2+…O2− [14] | 683.5 | 0.3332 | 0 | 2 | 8.77 |
| Pb2+…O2− [13] | 72276.42 | 0.2223 | 0 | −4 | 172.7 |
| Sr2+…O2− [14] | 1400 | 0.35 | 0 | 1.33 | 21.53 |
| Cu2+…O2− [15] | 3799.3 | 0.2427 | 0 | 2 | 99999 |
| Nd3+…O2− [16] | 1379.9 | 0.3601 | 0 | 3 | 99999 |
| Bi3+…O2− [13] | 49529.35 | 0.2223 | 0 | −5.51 | 359.55 |
| Y3+…O2− [16] | 1345.1 | 0.3491 | 0 | 3 | 99999 |
| Yb2+…O2− [16] | 1309.6 | 0.3462 | 0 | 3 | 99999 |
| Sc3+…O2− [16] | 1299.4 | 0.3312 | 0 | 3 | 99999 |
| Al3+…O2− [14] | 1114.9 | 0.3118 | 0 | 3 | 99999 |
| Ti4+…O2− [17] | 877.2 | 0.38096 | 9 | −35.86 | 95 |
| Zr4+…O2− [18,19] | 985.869 | 0.376 | 0 | 1.35 | 169.617 |
| Ce4+…O2− [18] | 1986.83 | 0.3511 | 20.4 | 7.7 | 291.75 |
| Sn4+…O2− [16] | 1056.8 | 0.3683 | 0 | 1.58 | 2037.8 |
| Nb5+…O2− [20] | 1796.3 | 0.34598 | 0 | −4.497 | 1358.58 |
| Mo6+(W6+)…O2− [13,21] | 767.43 | 0.4386 | 0 | 5.89 | 7.69 |
The calculated “solution energies” are given in Table 5. For trivalent cations, except Al, negative solution energies were obtained for dopant in the La site, which is in good agreement with the existence of compounds, such as: Nd3TaO7, Yb3TaO7, Y3TaO7, Bi3TaO7 and Sc3TaO7 [3,23]. In contrast, substitution for tantalum by trivalent cations is not favorable, since the solution energy is positive. The same trend is evidenced for tetravalent cations, excepted a few differences: dopants with intermediate ionic radii, such as Sn or Zr, seem to incorporate more easily into the La(2) site. The Ta site is more easily substituted by pentavalent or hexavalent cations, which is not surprising, as the compounds La3NbO7, La3MoO7 and La3MoO7.5 with the same structure do exist [24,25,26].
Solution energies calculated on La and Ta sites (the ionic radius of cation in 6-fold coordination from Shannon [22] is given for information).
| Ionic radius (Å) | Esol(La1) | Esol(La2) | Esol(Ta) | |
| Ca2+ | 1.00 | 0.64 | 0.70 | 17.28 |
| Mg2+ | 0.72 | 3.84 | 3.27 | 17.91 |
| Ni2+ | 0.69 | 4.17 | 3.59 | 18.17 |
| Pb2+ | 1.19 | −0.66 | −0.01 | 19.93 |
| Sr2+ | 1.18 | −0.23 | 0.26 | 18.24 |
| Cu2+ | 0.73 | 4.48 | 3.56 | 17.33 |
| Nd3+ | 0.98 | −2.11 | −2.30 | 11.42 |
| Bi3+ | 1.03 | −1.63 | −1.95 | 12.53 |
| Y3+ | 0.89 | −1.45 | −2.07 | 10.65 |
| Yb3+ | 1.02 | −1.16 | −1.92 | 10.44 |
| Sc3+ | 0.74 | 0.08 | −1.09 | 10.16 |
| Al3+ | 0.53 | 3.05 | 1.68 | 11.38 |
| Ti4+ | 0.60 | 2.09 | 0.38 | 4.93 |
| Zr4+ | 0.74 | 1.05 | −0.61 | 4.44 |
| Ce4+ | 0.87 | 0.80 | 0.07 | 7.09 |
| Sn4+ | 0.69 | 1.03 | −0.74 | 4.18 |
| Nb5+ | 0.64 | 3.03 | 0.40 | −0.57 |
| Mo6+/W6+ | 0.59/0.6 | NC | NC | 0.66 |
Partial substitution of La3+ and Ta5+ with an element of valence higher than 3 and 5, respectively, will favour an interstitial oxide ion migration mechanism (an oxide ion will migrate from an interstitial site to another). However, such a mechanism supposes interstitial sites to be close to each other, which is not the case. Since the cavities in the structure are spaced by at least 3.4 Å, an interstitial oxide ion migration pathway is therefore unlikely to occur and a vacancy oxide ion migration mechanism (an oxide ion from its crystallographic site will migrate to a vacancy, induced by the substitution) must be preferred. Such a mechanism will be possible after substitution of either La3+ ion by a divalent cation or Ta5+ by di, tri or tetravalent cations. The calculations show that the latter substitutions are energetically unfavorable, whatever the valence of the substituting cations. Only the former substitution may be energetically favorable.
At the end, Sr and Pb with negative solution energies appear as the most promising substituent for the La site. Moreover, they would prefer the La(1) site. This result is in good agreement with the possibility of partial substitution for La with Sr in La3NbO7 as reported by Shimura et al. [25]. In contrast, Ca and Mg would not be favorable. However, the value obtained for Ca is low and one must be careful with these values of solution energy, since the La3−xCaxTaO7 solid solution was recently studied by Haugsrud and Risberg [27]. Because of the lack of accuracy on the interatomic potentials, one must keep in mind that these calculations just give an order of magnitude and must be interpreted with care.
Pb being reducible in the condition of use of a fuel cell, it was not further considered in this study and, based on these calculations, it was decided to focus on the La3−xSrxTaO7−x/2 solid solution.
3.3 Evidence of a La3−xSrxTaO7−x/2 solid solution
X-ray diffraction pattern and Raman spectra corresponding to La3−xSrxTaO7−x/2 with x = 0, 0.05, 0.10, 0.15, 0.20, 0.25, 0.30 are given in Figs. 2 and 3, respectively. A shift of Bragg peaks is clearly evidenced for substitution ratio up to x = 0.15. A plateau is then to be noticed. This evolution is confirmed by Raman spectroscopy. The features of the spectra are similar for x = 0 up to x = 0.15 and no significant modification is observed in the very low wave numbers (lattice vibrations): this indicates no phase transition. The observed band at 725 cm−1, assigned to the stretching motion of the Ta-O bond, shifts towards the high frequencies when the substitution ratio increases, which characterizes the existence of a solid solution. For substitution ratio higher than 0.15, an extra band is observed at 766 cm−1, which confirms a solid solution domain for 0 ≤ x < 0.20.
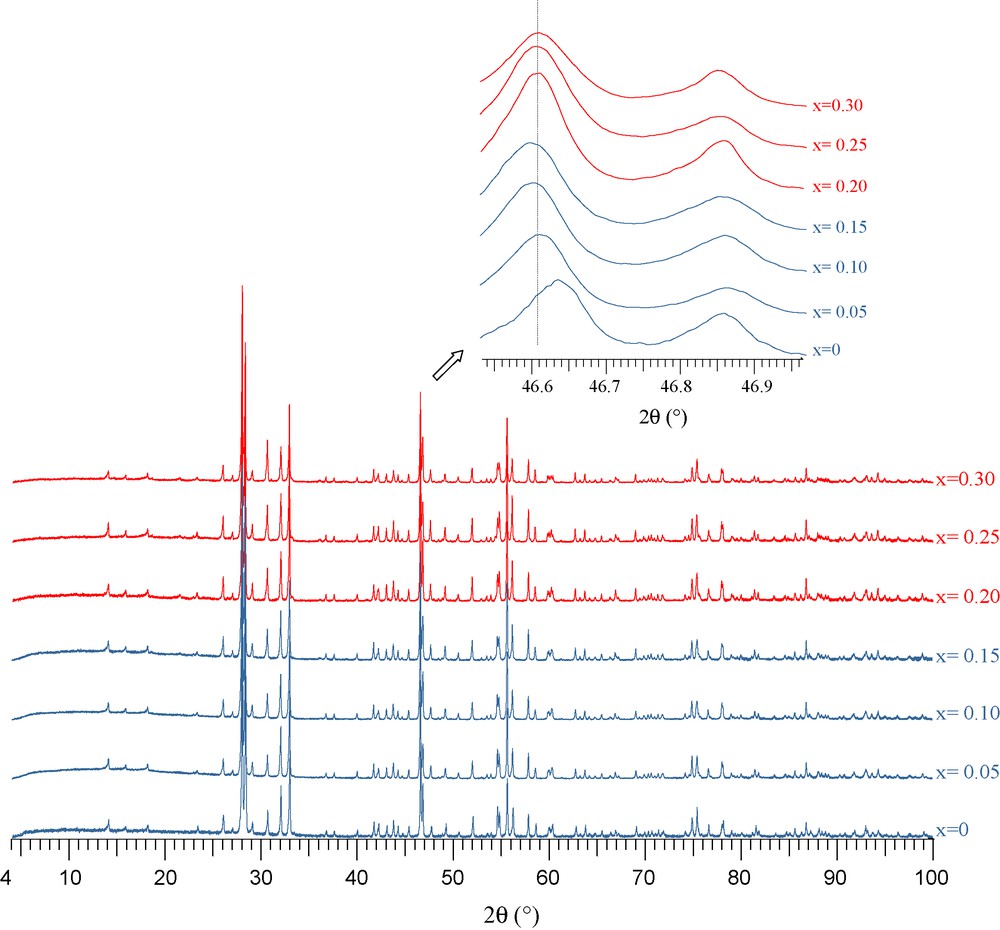
X-ray diffractograms corresponding to La3−xSrxTaO7−x/2, x = 0, 0.05, 0.10, 0.15, 0.20, 0.25, 0.30.

Raman spectra for La3−xSrxTaO7−x/2, x = 0, 0.05, 0.1, 0.15, 0.20, 0.25, 0.30.
From X-ray diffraction data, unit cell parameters were refined. Their evolution is given in Fig. 4. An increase of the b unit cell parameter with the substitution ratio is clearly observed with a plateau for substitution ratio higher than 0.20.

Evolution of unit-cell parameters and unit cell volume in function of the strontium ratio.
With the aim of characterizing the effect of partial substitution on the oxide ion conduction, impedance spectroscopy was performed on the parent compound and on composition x = 0.10. Dense pellets, 8.5 mm in diameter, 1.8–2 mm in thickness, were prepared. To decrease the particle size, powders were attrition milled prior to sintering. Ceramics with relative density higher than 97–98% were obtained by SPS. Within the experimental error, a good reversibility of Arrhenius plots was observed between heating and cooling and the two cycles. Therefore, only conductivity obtained during the second cooling is reported in Fig. 5. To verify the stability of the sample response, impedance spectra were measured repeatedly with a 1% drift tolerance and five successive measurements at maximum. Although good stability was obtained for a temperature higher than 300 °C, a 2% deviation was to be noticed at lower temperature. Experiments were carried out in air (static atmosphere) but moisture was not controlled and this lower stability could be due to a small interaction with water at low temperature. The Arrhenius plots of both compositions are compared to the most commonly used electrolyte for SOFCs, YSZ [28] (Fig. 5). The conductivity remains lower than YSZ, but it is worth noting, it is increased by more than one order of magnitude at 700 °C when strontium is introduced into the structure. Whereas a relatively high activation energy is observed for La3TaO7 (1.44 eV), it is about 0.9 eV, in the same range as YSZ, for the doped compound.

La3TaO7 and La3−xSrxTaO7−x/2, x = 0.1 Arrhenius plots of conductivity, compared to YSZ.
4 Conclusions
The aim of this article was to illustrate the approach our group considered for the research of new oxide ion conductors, which could be suitable as electrolyte for SOFCs. By defect chemistry, partial substitution of the main element with cations with different valence, one can expect to introduce oxygen vacancy or oxygen interstitials, which should modify the oxygen transport in such ceramics. Here, we focus on La3TaO7 whose crystalline structure was favorable to oxide ion diffusion. Atomistic simulations were used as a first approach to define possible defects, being aware that energy interactions are simplified and one must be careful with the accuracy of calculated energy values. As expected, partial substitution of La with an element of valence III, such as Sc, Nd, Y, Yb, Bi, is favorable; on the Ta site, substitution with dopant of valence V or VI is the most favorable. However, because of the space between cavities in the structure, an interstitial oxide ion migration mechanism in this system is unlikely and a vacancy oxide ion migration mechanism should be preferred. The introduction of dopant with lower valence in the Ta site does not seem possible. From the calculation, introduction of cation of valence II was more likely in the La site. A La3−xSrxTaO7−x/2 solid solution was indeed evidenced for x up 0.15 and it was shown that the conductivity of the parent compound was increased by more than one order of magnitude at 700° C when 3.33% strontium was introduced in the La site. The moisture of the atmosphere was not perfectly controlled but a good reversibility on heating and cooling was observed for the two samples. Since proton conduction was recently observed for La3NbO7 and La3−xCaxTaO7 [27], one cannot exclude the possibility of proton conduction in these materials. Impedance spectroscopy and TGA under wet atmosphere are currently under progress to verify this assumption. Recently, to check the stability of these compounds, we measured conductivity under variable oxygen partial pressure. At 800 °C, under hydrogen, a good stability was observed, but a slight increase of conductivity with oxygen partial pressure was also to be noticed, indicating a p-type semi-conduction character. In this study, we demonstrate the possibility to increase oxide ion conduction by partial substitution of cation in a structure favorable for oxide diffusion. However, ionic transport in Weberite derivatives is likely more complicated than only oxide ion diffusion and further work is in progress for a better understanding of their transport properties.
Acknowledgements
The authors are very grateful to the Region Nord Pas de Calais and Picardie and the CNRS for N.P. funding.