

1 Introduction
The impressive advances in electronics have placed significant demand on micropower sources, and rechargeable Li-ion microbatteries based on thin films are promising candidates to power such electronic devices as “smart” cards, RFID tags, remote sensors, power implantable medical devices, etc. [1–3]. However, the electrochemical performances of ultra thin all-solid-state batteries are limited because only 2D thin films are employed as anode, electrolyte and cathode materials. In general, the total thickness of the stacking films is in the range of 10 to 15 μm and the resulting battery, which is formed of parallel plates, reveals relatively low power and energy densities. In order to ensure significant advances for extended applications, it is crucial to improve the electrochemical performance by investigating new electrode and electrolyte materials as well as innovative manufacturing processes. Recently, better rate capability, capacity and cycling behaviour have been observed for nanostructured electrodes (e.g. Ortiz and Tirado [4]) and especially self-organized titania nanotubes (TiO2nt) [5–10]. The fabrication of self-organized TiO2nt using several procedures has been widely reported for different kinds of applications [11–15].
So far, the electrochemical characteristics of such nanostructured electrodes have been reported only for liquid organic electrolytes (e.g. LiPF6 (EC:DEC)), which are not applicable for the fabrication of all-solid-state microbatteries. Besides this, the thin-film deposition technologies currently employed for depositing the solid electrolyte (e.g. lithium phosphorus oxynitride [LiPON]) [2,15–19] are not adequate to achieve the total or conformal filling of the nanotubular electrodes. Indeed, all conventional top-down approaches cannot be used since gradual accumulation of materials at the top of the electrode closes the nanotube openings during the deposition process. Thus investigating bottom-up approaches such as electrodeposition is a convenient way to circumvent this problem and increase the electrode/electrolyte interface for the fabrication of micropower sources with enhanced electrochemical performance. However, the glassy solid electrolytes that are currently employed cannot be obtained by electrochemical synthesis.
Recently, polymer electrolytes have also been demonstrated to be promising candidates as solid electrolytes for lithium batteries. Particularly, significant progress has been achieved with poly(ethylene oxide)-functionalized methyl methacrylate (PEO-PMMA) copolymers blended with PVDF, leading to a polymer electrolyte with high ionic conductivity (2.79 × 10−3 S.cm−1) at room temperature and high electrochemical stability (5.0 V vs Li/Li+) [16]. A large variety of PEO-based copolymers are studied for electrolyte applications in conventional lithium batteries and for microbatteries as well. However, these sort of electrolytes have solely been developed for large batteries because both conventional and alternative synthesis procedures (e.g. thick coating by dispersion/evaporation, thermal curing and phase inversion technique) [16–18] lead to the formation of thick layers (> 100 μm) which are not compatible with the limited size of microbatteries (maximum 15 μm thickness).
The goal of this work is to propose a reliable, versatile, and economically viable procedure to grow copolymer electrolyte thin films (< 1 μm) on different electrodes and especially nanostructured electrodes. In complement with the preliminary results recently reported [19], direct electropolymerization of PEO-MMA into the titania nanotubes is given an in-depth study. Particularly, the electrosynthesized polymer electrolyte is characterized by several techniques and we show that the resulting electrode/electrolyte interface leads to significant improvement of the electrochemical performance compared to systems based on the use of solid electrolytes deposited by top-down techniques.
2 Experimental
In this work, Ti foils, supplied by Sigma-Aldrich, with a thickness of 0.127 mm and 99.7% purity were cut and cleaned by sonicating in acetone, isopropanol and methanol during 30 min. The substrates were then rinsed with distilled water, and dried with compressed air. The electrochemical anodization of the Ti foils was carried out by potentiostatic experiments in 1 M H3PO4 + 1 M NaOH + 0.4 wt % HF electrolyte at room temperature. An electrochemical cell with a two-electrode arrangement was used. The working electrode was a piece of titanium (1.0 cm × 1.0 cm) and a platinum grid of large surface area served as counter electrode. The anodization experiments consisted of applying a constant voltage of 20 V during 2 hours using an EG&G PARSTAT 2273 potentiostat/galvanostat. The anodized Ti samples were immediately rinsed with distilled water and dried with compressed air. The as-formed titania nanotube layers were then used as working electrodes for electropolymerization by cyclic voltammetry, which involved sweeping at 25 mV/s from 0 V to −2.5 V with a reverse scan using saturated calomel electrode (SCE) as reference. Cathodic polarization curves were recorded using EG&G PARSTAT 2273 potentiostat/galvanostat. The electrolyte was a 0.035 M LiN(CF3SO2)2, so-called LiTFSI, aqueous solution. This salt is the prototype of a new family of bulky lithium salts with bulky anions combining great charge delocalization favourable for ionic dissociation in a solvating polymer such as PEO, good chemical, electrochemical and thermal stabilities and also a plasticizing effect which decreases the crystallinity of the host polymer, enhancing ionic mobility. This LiTFSI salt with 99.0% purity was used as received from Sigma-Aldrich.
After purging the electrolyte solution with nitrogen gas for 10 minutes, 2 g of PEO-functionalized methyl methacrylate monomers (MMA-(PEO)n with n = 475 used as received from Aldrich) was added. After electropolymerization, the samples were dried in an oven at 60 °C to remove part of the residual water content of the polymer. The morphological studies and the chemical analysis of the samples were carried out using a Philips XL-30 FEG scanning electron microscope (SEM) equipped with an energy dispersive spectroscopy (EDS) unit. FTIR spectroscopy in the range of 4000 to 400 cm−1 was performed using Equinox 55 Brucker in the diffuse reflectance mode.
1H NMR spectra were obtained with a Bruker Advance DPX 400 MHz spectrometer at room temperature in 5 mm o.d. tubes. The 1H chemical shifts were referenced to the solvent peak: CDCl3 (7.26 ppm). The determinations of polymer molecular weights and molecular weight distribution were performed on a system composed of a Waters 717 plus auto-sampler, a Waters 600 system controller and a Waters 600 fluid unit and two detectors: a waters 2414 differential refractometer and a UV/visible detector Waters 486, both used at 40 °C. THF + 0.25%vol toluene was used as eluent at a flow rate of 0.7 mL.min−1 after filtration on Alltech nylon membranes with a porosity of 0.2 μm. The column oven was kept at 70 °C, and the injection volume was 20 μL. Two ResiPore columns (600 mm, 7.5 mm) (Polymer Laboratories) were used in series. The system was calibrated using PS standards (Standards Varian Easical PS-2) in the range 1000–400 000. Electrospray Ionization mass spectrometry (ESI-MS) were performed with a 3200 QTRAP (Applied Biosystems SCIEX) mass spectrometer.
The electrochemical measurements were done using Li/LiPF6(EC:DEC)/PEO-PMMA(LiTFSI)/ntTiO2 cells. The Whatman glass microfiber, which acted as a separator, was soaked with the liquid electrolyte, and the titanium metal substrate served as the current collector for the TiO2nt. No additives such as polyvinylidene fluoride (binder) or carbon black (conductive agent) were required. The electrodes were previously dried at 60 °C and the coin cells were assembled in a glove box filled with argon and having moisture and oxygen contents less than 2 ppm. The lithium coin cells were galvanostatically cycled using a VMP Biologic Science Instrument in the voltage range of 0.05 to 2.6 V and current densities of 14 (C/5), 70 (1C) and 350 μA/cm2 (5 C).
For comparison purposes, LiPON thin films were deposited onto as-prepared TiO2nt by rf magnetron sputtering of an Li3PO4 target in a pure N2 atmosphere with a BAK550 (Balzers) apparatus. The base pressure was 2 × 10−4 Pa. Plasma ignition was performed with argon before introduction of nitrogen. Prior to the deposition of thin films, a presputtering of 30 min was performed. The target of 7.5 cm diameter was prepared either by hot pressing or uniaxial pressing (at room temperature) of a commercial Li3PO4 powder (Aldrich, 99.9%).
3 Results and discussion
Electrochemical polymerisation (ECP) is a powerful technique for depositing thin passive films onto materials for corrosion protection and adhesion enhancement. It is also widely adopted to produce highly pure polymer films for applications in electrocatalysis, micro-optoelectronics, and photo-electrochemistry [20–26]. Depending on the role of the working electrode anode or cathode, the electropolymerization mechanism can be cationic, anionic, or free radical. The polymerization process can be initiated by the monomer or by added initiators when the monomer is not electrochemically active. In the present case, electropolymerization is expected to occur according to the free radical mechanism proposed by Cram et al. [25] (a reaction mechanism is given in Scheme 1). The reduction of H+ to produce H2 is accompanied by the formation of H• radicals that can react with the monomers, leading to the polymerization of methyl methacrylate. The possible free radical-induced electropolymerization mechanism is confirmed through the examination of the cyclic voltammograms given in Fig. 1. Actually, the similar general shape of both polarization curves indicates that only the reduction reactions of H+ and TiO2 take place within the studied potential window. In accordance with the Nernst equation, the increasing cathodic current starting at around -0.66 V/SCE is attributed to the reduction of H+ and the production of H• radicals, which are responsible for the polymerization process. At −1.22 V/SCE applied potential, it can be observed the reduction of Ti4+ into Ti3+ ions with its counter reaction on the reverse scan as has been reported in literature [27]. At more cathodic potentials, H2 evolution becomes predominant. The main difference between these two voltamogramms is the kinetics of the reactions. Indeed, when the polarization is performed in the monomer-containing electrolyte, lower current densities are recorded on both scans. This result clearly shows the formation of an insulator layer hindering the redox reactions.

Electropolymerization of PEO-functionalized methyl methacrylate based on the free radical mechanism.
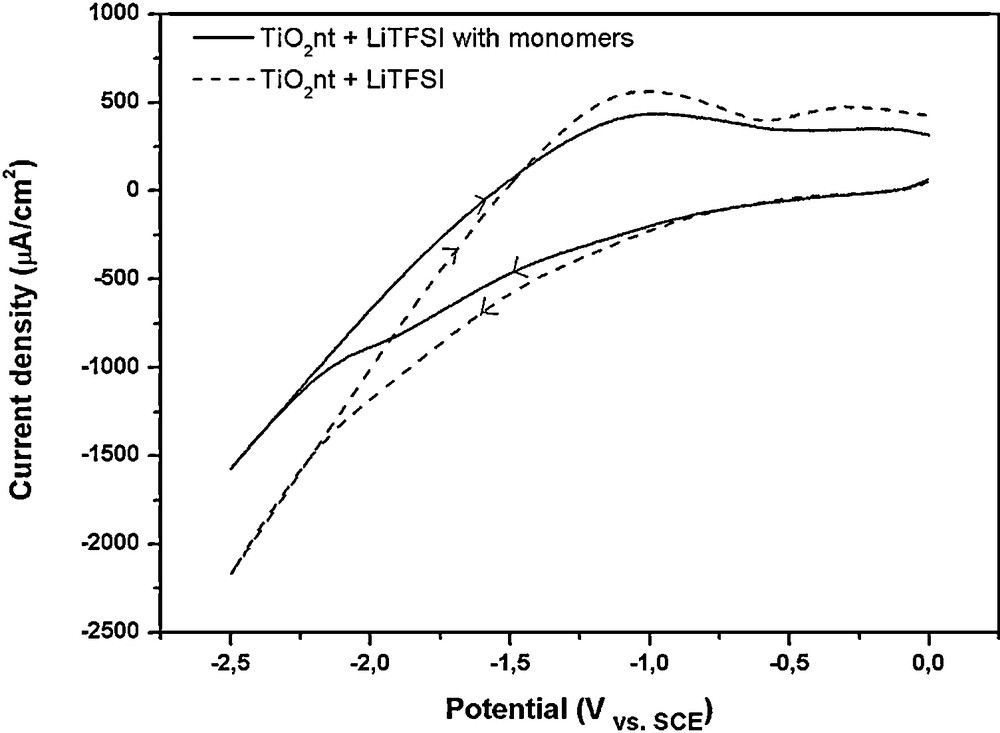
Cyclic voltamogramms recorded between 0 V and −2.5 V vs SCE of TiO2nt layer in 0.035 M LiTFSI aqueous solution, and in 0.035 M LiTFSI aqueous solution + 2 g PEO−functionalized MMA. Sweeping rate of 25 mV/s [19].
The deposition of the copolymer into the titania nanotube layer is confirmed by morphological and chemical analysis. The SEM image of as-formed titania nanotubes that are 700 nm long with diameters of around 80 nm can be compared with the polymer-embedded titania nanotube layer (Fig. 2a, b). Apparently, the nanotubes are completely filled by a thin polymer film, suggesting that the electrodepositon begins from the bottom of the nanotubes [26,28]. A fine control of the copolymer thickness can be achieved because the growth mechanism depends on the monomer concentration and on the bath immersion time once the free radicals are formed.

SEM images of (a) as-formed TiO2nt, (b) copolymer-embedded TiO2nt, and (c) TiO2nt with 200 nm of LiPON deposit.
The EDS spectra shown in Fig. 3 depict very high carbon content for the polymer-embedded nanotubes. The source of the carbon was also clearly identified to be the copolymer by the FTIR spectroscopy results shown in Fig. 4. Indeed, PEO and PMMA bands can be easily identified (see the corresponding band values obtained from literature in Table 1) [20,21,27,29–31]. It can be noted that no such bands are present in the FTIR spectrum obtained from the as-prepared TiO2nt sample, which displays just the Ti-O-Ti stretching mode of TiO2 at less than 1000 cm−1.

EDS spectra of (a) as-formed TiO2nt and (b) copolymer-embedded TiO2nt.

FTIR spectra of (a) copolymer-embedded TiO2nt and (b) as-formed TiO2nt.
Typical FTIR peak positions of PMMA-b-PEO.
| Peak position (cm−1) | Peak assignment |
| 2880 | CH stretching vibration in PMMA |
| 1730 | CO carbonyl stretching group in PMMA |
| 1640 | Adsorbed water assigned to the δHOH bending mode |
| 1460 | CH2 asymmetric bending in PEO |
| 1346–1360 | CH2 wagging in PEO |
| 1300 | OCH3 stretching vibrations in PMMA |
| 1250–1280 | CH2 twist |
| 1200–1212 | CO stretching vibrations |
| 1147–1150 | CO stretching vibrations |
| 1104 | COC stretching vibrations in PEO |
| 850 | CH2 rocking in PEO |
The generation of the copolymer by electropolymerization was confirmed by 1H NMR experiments. The high solubility of the resulting PEO-PMMA in water enabled its removal from the nanotubes by dissolution and further analysis in both organic and aqueous solvents were then carried out. The 1H NMR spectra were recorded in CDCl3. Examination of the 1H NMR spectrum of the polymer evidences the disappearance of the characteristic set of peaks of the vinylic group located at 5.57 and 6.12 ppm respectively, therefore demonstrating that a reduction of the methylmethacrylate group had occurred (Fig. 5). Formation of the copolymer is also corroborated by the strong shift observed for the methyl group of the methacrylic unit which shifts from 1.95 ppm to 1.48 ppm, thus confirming the polymerization and the formation of a saturated chain. Broadening of all the signals is also in agreement with the formation of a polymer, this phenomenon being typically observed for the NMR spectra of polymers. Finally, careful analysis of the 1H NMR spectrum evidences the formation of copolymers of various lengths by the presence of several signals located between 4.0 and 4.6 ppm and assigned to the CH2 group of the ester. To verify that the polymeric material was not issued from a simple electrochemical reduction of the methacrylic group, a comparison with the 1H NMR spectrum of a model ester molecule (ethyl isobutyrate) that bears an isopropyl group was carried out (Fig. 6).

1H NMR spectra of (a) the monomer and (b) the polymer.
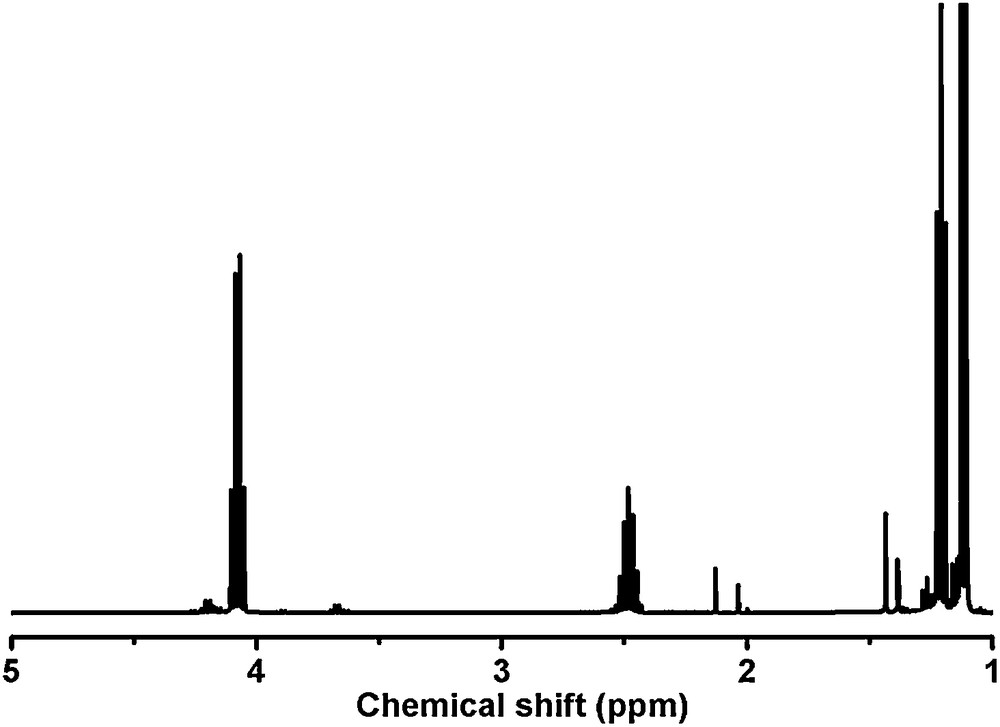
1H NMR spectrum of the ethyl isobutyrate recorded in CDCl3.
Comparison of NMR spectra shown in Figs. 5b and 6 clearly indicates that the septuplet of the CH group at 2.48 ppm and the doublet of the CH3 groups at 1.12 ppm of ethyl isobutyrate were not observed on the NMR spectrum of the electropolymerized material, therefore providing another proof for the formation of the desired polymer. Further evidence of polymerization was provided while comparing our NMR spectrum with the 1H NMR spectra of PEO-PMMA copolymers synthesized by various techniques such as Atom Transfer Radical Polymerization (ATRP), [32,33] photopolymerization, [34] anionic polymerization [35] and free radical polymerization [36]. In each case, a good agreement was determined between the chemical shifts reported in the literature for both the CH3 and the CH2 groups of the PMMA chains and the chemical shift of the same groups for the electropolymerized material.
The polymeric material was also investigated by Size Exclusion Chromatography (SEC), applying a Refractive Index (RI) detector and Polystyrene (PS) standards. Comparison of the SEC traces of both the monomer and the electropolymerized material confirmed the formation of a copolymer (Fig. 7). Although the retention time of the polymeric material is close to that of the monomer, a clear broadening of the trace with a shorter retention time compared to the monomer can be observed indicating the formation of polymers. As the electropolymerization does not induce significant changes in the retention time, formation of oligomers instead of polymers is also evidenced. No traces of high molecular weight copolymers are even detected by SEC. As other evidence for the formation of oligomers, a number average molar mass (Mn) of about 510 g.mol−1 and a polydispersity index (PDI) of 1.846 were determined as the characteristics of the electropolymerized material using PS standards. Oligomers with chains comprising a few monomer units are thus produced by electropolymerization and the high value of the PDI is consistent with a non-controlled polymerization process.

SEC traces of the monomer (solid line) and the polymeric material (dotted line). Eluent system: THF + 0.25%vol toluene as the internal reference.
Formation of oligomers was confirmed by Electrospray Ionization (ESI) mass spectrometry experiments. Mass spectrometry is a powerful technique for characterizing the chemical compositions and the molar mass distributions of polymers while inducing little or no fragmentation when a soft-ionization method such as ESI-MS is used [37]. Elucidation of a polymer structure, characterization of a polymer end-groups and determination of the mass of repeat units can also be achieved. In the limit of the complexity of the mass spectrum (Fig. 8), a large number of oligomeric species led to overlapping peaks. The complexity of the ESI-MS spectrum of the electropolymerized material may also originate from the initial polydispersity of the monomer used during the polymerization process. In the present case, ESI-MS analysis of the commercially available monomer evidenced a broad distribution of monomers comprising at least thirteen different oligomers. In contrast to SEC, ESI-MS also enables to determine the absolute molecular weight and molecular weight distribution. Four distinct ion series were identified in the ESI-MS spectrum as ammonium adducts [M + NH4]+. Fig. 8 shows the ESI spectrum obtained in the positive ion mode and the peak data of the four ion series are given in Tables 2a–d. The spacing of 1 Da between ion peaks indicated the presence of only singly charged species. Considering the general formula for the electropolymerized material Cp(2q+5)Hp(4q+8)Op(q+2)XY, with “p” representing the number of MMA repeating units, “q” the length of the ethyleneglycol chain, “X” and “Y” the two end-groups, oligomers comprising between one to four repeating MMA units were detected. However, the presence of higher molecular weight oligomers in the electropolymerized material cannot be excluded, the intensity of the “molecular ion” generally decreasing with increasing oligomer sizes [38]. Due to the dependence of the ionization efficiency on the molecular weight, biased molecular weight distribution could be deduced. Based on the abundance of the mass-to-charge ratios, number average molecular weights (Mn) of 563, 527, 549, and 537 g.mol−1 with respective PDI of 1.04, 1.04, 1.03, 1.03 could be determined for the four distributions of oligomers.

ESI spectrum of (a) monomers and (b) electropolymerized material obtained in the positive ion mode.
ESI-MS peak m/z values of the first polymer distribution.
| q | p | |||
| 1 | 2 | 3 | 4 | |
| 1 | 226 | 370 | 514 | 658 |
| 2 | 270 | 414 | 558 | 702 |
| 3 | 314 | 458 | 602 | 746 |
| 4 | 358 | 502 | 646 | 790 |
| 5 | 402 | 546 | 690 | 834 |
| 6 | 446 | 590 | 734 | |
| 7 | 490 | 634 | 778 | |
| 8 | 534 | 678 | 822 | |
| 9 | 578 | 722 | 866 | |
| 10 | 622 | 766 | ||
| 11 | 666 | 810 | ||
| 12 | 710 | 854 | ||
| 13 | 754 |
ESI-MS peak m/z values of the second polymer distribution.
| q | p | ||
| 1 | 2 | 3 | |
| 1 | 254 | 398 | 542 |
| 2 | 298 | 442 | 586 |
| 3 | 342 | 486 | 630 |
| 4 | 386 | 530 | 674 |
| 5 | 430 | 574 | 718 |
| 6 | 474 | 618 | 762 |
| 7 | 518 | 662 | 806 |
| 8 | 562 | 706 | 850 |
| 9 | 606 | 750 | |
| 10 | 650 | 794 | |
| 11 | 694 | 838 | |
| 12 | 738 | ||
| 13 | 782 |
ESI-MS peak m/z values of the third polymer distribution.
| q | p | ||
| 1 | 2 | 3 | |
| 1 | 268 | 412 | 556 |
| 2 | 312 | 456 | 600 |
| 3 | 356 | 500 | 644 |
| 4 | 400 | 544 | 688 |
| 5 | 444 | 588 | 732 |
| 6 | 488 | 632 | 776 |
| 7 | 532 | 676 | 820 |
| 8 | 576 | 720 | 864 |
| 9 | 620 | 764 | |
| 10 | 664 | 808 | |
| 11 | 708 | ||
| 12 | 752 | ||
| 13 | 796 | ||
| 14 | 840 |
ESI-MS peak m/z values of the fourth polymer distribution.
| q | p | ||
| 1 | 2 | 3 | |
| 1 | 284 | 428 | 572 |
| 2 | 328 | 472 | 616 |
| 3 | 372 | 516 | 660 |
| 4 | 416 | 560 | 704 |
| 5 | 460 | 604 | 748 |
| 6 | 504 | 648 | 792 |
| 7 | 548 | 692 | 836 |
| 8 | 592 | 736 | |
| 9 | 636 | 780 | |
| 10 | 680 | 824 | |
| 11 | 724 | 868 | |
| 12 | 768 | ||
| 13 | 812 |
Fig. 9 shows the volumic capacity vs cycle number for the as-formed TiO2nt and copolymer-embedded TiO2nt at two kinetic rates. Due to the gel-like nature of the polymer, no heating was required for the electrochemical studies. It clearly appears that the electrochemical performances of the hybrid system summarized in Table 3 are promising for microbatteries. The capacity fading is minimal even with 50 cycles, showing that the electrodeposited copolymer electrolyte can bear the volume variations of the host electrode during cycling. Compared to the as-formed TiO2nt, at current densities of 14 μA/cm2 (C/5) and 70 μA/cm2 (1C), the copolymer-embedded TiO2 nanotubes reveal higher capacity values (∼8%).

Capacity vs cycle number for the as-formed and copolymer-embeddded TiO2nt cycled at 1C and C/5 [19].
Values of capacities and efficiency on cycling of as-prepared TiO2nt and copolymer-embedded TiO2nt in experimental test cells at different kinetics.
| Electrode | Kinetic rate | 1st DC | 1st RC | IC | 50th C | E (%) |
| TiO2nt | C/5 | 176 | 75 | 101 | 60 | 80 |
| 1C | 136 | 66 | 70 | 50 | 76 | |
| TiO2nt + copolymer | C/5 | 189 | 83 | 106 | 65 | 78 |
| 1C | 155 | 70 | 85 | 54 | 77 |
Moreover, for the same current condition (350 μA/cm2), the electrochemical cell based on the copolymer (gel-like) electrolyte has much higher capacity values than the system based on LiPON electrolyte (Fig. 10). This can be explained by the examination of the electrode/electrolyte interface. It can be seen from Fig. 2c that the LiPON deposit covers completely the top of the nanotubes and does not appear to enter the voids within or between the nanotubes, which is otherwise possible with the polymer electrolyte (Fig. 2b). It can be asserted that the good performance of the copolymer-embedded TiO2 nanotubes is not only as a result of the good conductivity of the polymer but also due to an improved electrode/electrolyte interface brought about by the proper filling of the nanotubes.

Capacity vs cycle number for copolymer-embedded TiO2nt and TiO2nt with 200 nm of LiPON deposit (cycled at j = 350 μA/cm2).
4 Conclusion
We report that electropolymerization is a viable technique to achieve the deposition of polymers into nanostructured electrodes. We have demonstrated that this bottom-up approach constitutes a promising alternative to other approaches currently developed for depositing polymer electrolytes. We can accurately control the thickness of the PMMA-b-PEO copolymer layer in the sub-μm scale while ensuring the total filling of self-organized titania nanotubes. We have shown that such a very thin copolymer electrolyte (< 1 μm) is adequate for bearing the volume variation of the host electrode during cycling. Furthermore, the high electrode/electrolyte interface combined with the remarkable properties of the copolymer electrolyte led to improved electrochemical performances. Improvement in capacity of 8% can be obtained. This strategy can be extended to other 3D nanostructured electrodes made of materials showing higher lithium ion storage capacity than titania (e.g. nanostructured Si). Hence, the electrochemical synthesis can be also transposed to a wide range of PEO-based monomers (e.g. PEO-functionalized styrene). Finally, this process is also compatible with the integrated circuit technology, opening the path towards the development of a fully integrated system for on-chip and flexible 3D microbatteries prepared solely by electrochemical techniques.
Acknowledgements
We acknowledge the French Ministry of Education, C-Nano PACA, region PACA and ANR JCJC no 2010 910 01 for financial supports.