

1 Introduction
Lectins, a superfamily of proteins, which recognize and bind to specific carbohydrate epitopes without modifying them, function as salient mediators of information transfer in biological systems, through the interaction with the glycans of glycoproteins, glycolipids and oligosaccharides [1]. Being involved in diverse aspects of cell sociology, lectins may be therapeutic targets or may be used to deliver drugs to their site of action [2,3]. Used in this particular case study, the galactoside-specific agglutinin from Viscum album L. (VAA; also named viscumin or MLI) is a heterodimeric (AB chains) toxic plant lectin that belongs to the family of ribosome-inactivating proteins (RIPs) of type II [4–6] that potently inhibits cellular protein synthesis. The A chain, responsible for the toxicity, has rRNA N-glycosidase activity and irreversibly damages eukaryotic ribosomes shutting down protein synthesis [7]. The B chain is a lectin that targets galactose-terminated glycans on the cell surface, enabling internalization of the toxic subunit subsequent to the docking process [8–11]. The toxicity of this potent biohazard akin to ricin can be neutralized by suitable inhibitors competing with the cell-surface glycans [12,13], which impede the attachment, and thus the internalization of the toxin into the target cells. This has made the toxin an attractive target for drug design in the search for new binding partners as potential inhibitors and also a robust test system for the respective studies [12–16].
Knowing that the binding of protein to a single carbohydrate moiety is often weak and that in physiological conditions carbohydrate–protein interactions usually benefit from multivalent carbohydrate epitopes engaged with multiple protein's carbohydrate recognition domains (CRDs) [17–19], a majority of the evaluated compounds (see library of compounds in Fig. 1) was designed with a substituted aromatic group at their aglyconic end, to favourably have an effect on the affinity and later be used in the preparation of multivalent glycoconjugates (by coupling the carboxy group with dentritic amines [20–23]). Details of the synthesis of the compounds studied are not yet published [24]. Multivalent enhancement has been seen for several lectins, as ricin [25] or human C-type lectins [26], to name a few. The main goal of this study was to define a strategy used for lectins suited to identify new synthetic monovalent ligands that could bind specifically to the VAA's CRD more strongly than lactose/galactose.
The library of compounds screened versus the target lectin.
The screening of this small library of novel galactose derivatives (Fig. 1) as VAA inhibitors also lead to new structural insights that can prove useful in the design of effective inhibitors for VAA activity. The study of potent lectin counter-receptors may provide not only a basis for the development of new pharmaceuticals, but also be valuable to offer insights into the protein–carbohydrate interaction mechanisms [27].
2 Results and discussion
STD NMR spectroscopy allows the detection and structural insights into ligand binding to large proteins, revealing information on vicinity of ligand protons to the protein [28]. Most noteworthy, and of great practical and experimental interest, there is no size limit for the protein receptor and no assignment of protein resonances is necessary. Also, there is no requirement for isotope labelling of either the protein or the ligand. Experiments require very small amounts of protein (< 20 μM) and an excess of ligand in fast exchange [29], in systems in which Kd values generally fluctuate between 10−3 to 10−7 M.
Herein, an experimental STD NMR competition-based protocol is applied to the study of the interaction of modified galactopyranosides with VAA. Besides validating the binding of the compound tested, it may also provide information on the specificity of two different ligands for the same active site of the receptor protein. Moreover, by using a reference ligand compound (termed “spy”, “reporter” or “probe” molecule, lactose (1), in the present study) with a known Kd value, it is possible to monitor the inhibitory potency of the ligand competing with the reference compound, by keeping track of the decrease of the reference STD signals (as shown in Fig. 2).
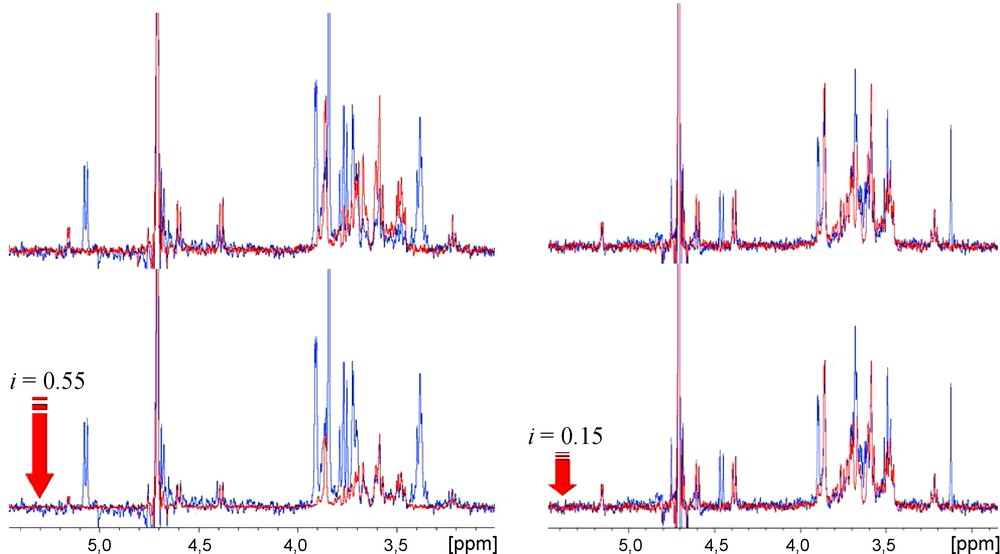
STD competition experiments: in red, STD spectrum of lactose in presence of VAA; in blue, STD spectrum of the same sample after introduction of the inhibitor (7, at the left; 9, at the right). It can be seen that the STD signal intensities of the reference compound lactose [present in a 4.6 mM concentration (100:1 ligand:protein ratio)] shows a higher (55%) or lower (15%) decreases after an equimolar introduction of inhibitors 7 or 9, respectively.
As an advantage, this competition method does not require the collection of several data points to determine an affinity constant, but it does require that the Kd and NMR signal response of the present “spy” molecule to be previously determined. This has been seen also in several 1D NMR studies with waterLOGSY [30], 1H transverse and selective longitudinal relaxation-filtered [31] (and STD [32–34]) experiments. In fact, STD NMR screening in competition mode, by monitoring the displacement of a well-characterized spy molecule by a test compound, eliminates the two major limitations of the ligand-based method: the false negatives of high affinity (on the low nanomolar range) test compounds, and the false positives resulting from limited test compound solubility and non-specific binding to the receptor.
The interference caused by each compound on the binding of lactose to VAA was followed and quantified by the decrease of lactose STD signals (see Fig. 2 as an example), and thus its inhibitory potency is calculated by the following equation [34]:
| (1) |
| (2) |
Inhibition constants as determined by STD-NM.
| Compound | Structure | Estimated KI (mM) |
| – | 0.870a [35] | |
| 1 | 0.813a [36] | |
| 2 | 0.91 ± 0.23 | |
| 3 | 0.80 ± 0.16 | |
| 4 | 1.05 ± 0.24 | |
| 5 | 1.09 ± 0.11 | |
| 6 | 1.10 ± 0.12 | |
| 7 | 0.56 ± 0.07 | |
| 8 | 0.41 ± 0.04 | |
| 9 | 4.11 ± 1.37 | |
| 10 | 2.19 ± 0.16 | |
| 11 | 1.30 ± 0.47 |
a Galactose and lactose values were taken from previous reports – by ITC – and should be mentioned in terms of Kd; lactose was chosen as the reference compound in the inhibitory analysis, due to the smaller overlap with the signals of the evaluated ligands than galactose; the range of error is associated with the deviation of an inhibition value i observed for different lactose protons.
As known from titration calorimetry, the interactions of VAA with lactose or galactose affinities values differ by only 0.4 kcal/M at 293.1 K [35,36]. From the results shown (Table 1) it can be seen that the introduction of aromatic aglycons (2, 3) on the galactose moiety had no influence on the affinity. Also, the inclusion of a nitrogen donor group at the phenyl ring's ortho position (4, 5) resulted in compounds with a range of affinity similar/slightly lower to that reported for lactose, with a minor increase of the inhibition value. Structural modifications made at the galactose C6 position, also similar to the results obtained for 4 and 5, revealed a small KI increase after the introduction of a linear azide group (6). In contrast, the replacement of the C6 hydroxyl group by a polar nitrogen group with hydrogen-bond donor proprieties resulted in increased affinity values (up to 2 × the value of lactose) for 7 and 8. Docking simulations were subsequently undertaken to help to rationalize the obtained data, making use of the X-ray structure of VAA available at the Protein Data Bank (PDB code 1PUM, protein complexed with galactose). Initial experiments were conducted with a β-D-galactose molecule in order to validate the docking simulation protocol. It was observed that the most populated cluster represented the docked molecule positioned at the same coordinates as the X-ray ligand (with exception for the hydroxyl group positioned at the C6, in a gt conformation relatively to the axial OH4), as seen in Fig. 3.
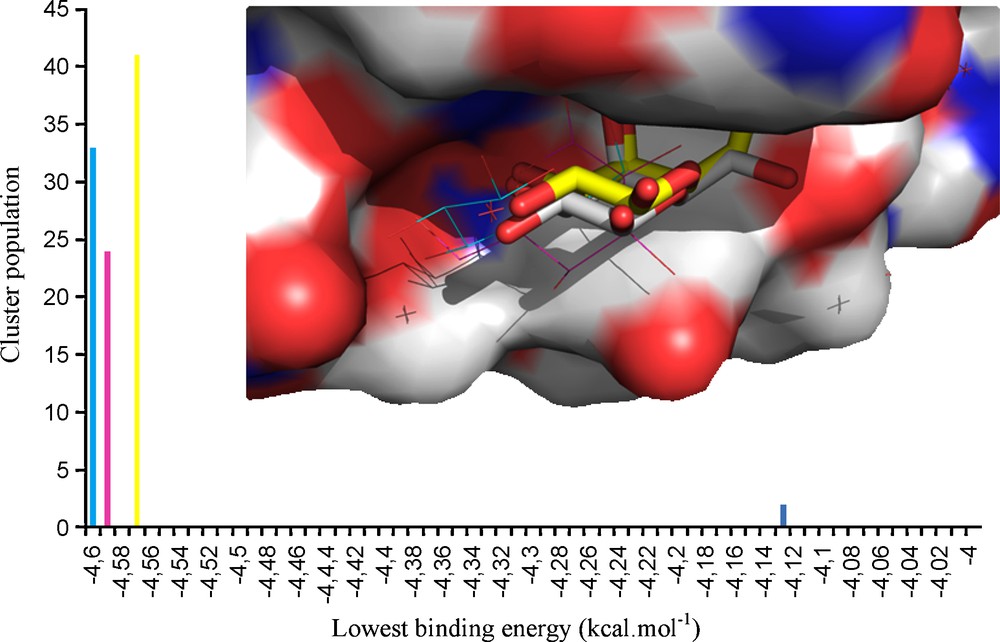
Representation of the Tyr CRD of viscumin (surface representation) complexed with galactose. Similar positioning of the galactose ligands is observed for both crystallographic (carbons displayed in grey) and docking-obtained structures (light blue and pink – shown in lines; yellow – shown in sticks; dark blue – 3D representation not shown). Hydrogen atoms are not shown for simplicity. The docking results can be seen in the clustering chart shown in the outer region of the figure.
Whenever working at VAA concentration values above 0.01 mg/ml, dimerization imposes steric restrictions at the Trp binding sites and subsequently reduces the net lactose binding, limiting its occupancy to the accessible Tyr site [37]. As for galactose, docking simulations of 7 and 8 were undertaken by using the same coordinates, and focusing on the Tyr site. By looking at the three-dimensional structure of the docked 7-VAA and 8-VAA complexes, it appears that the introduced amido and acetylamide groups could be involved in the formation of new intermolecular hydrogen bonds formed with the protein, namely with the Tyr249 residue (Fig. 3). These additional contacts seen in the docking simulations are consistent with the experimental values obtained by NMR. Both compounds 7 and 8 showed similar docking results for their most stable conformers (Fig. 4) and it was seen that the presence of the aromatic moiety seems to influence the position and orientation of the galactose ring of the docked structure.
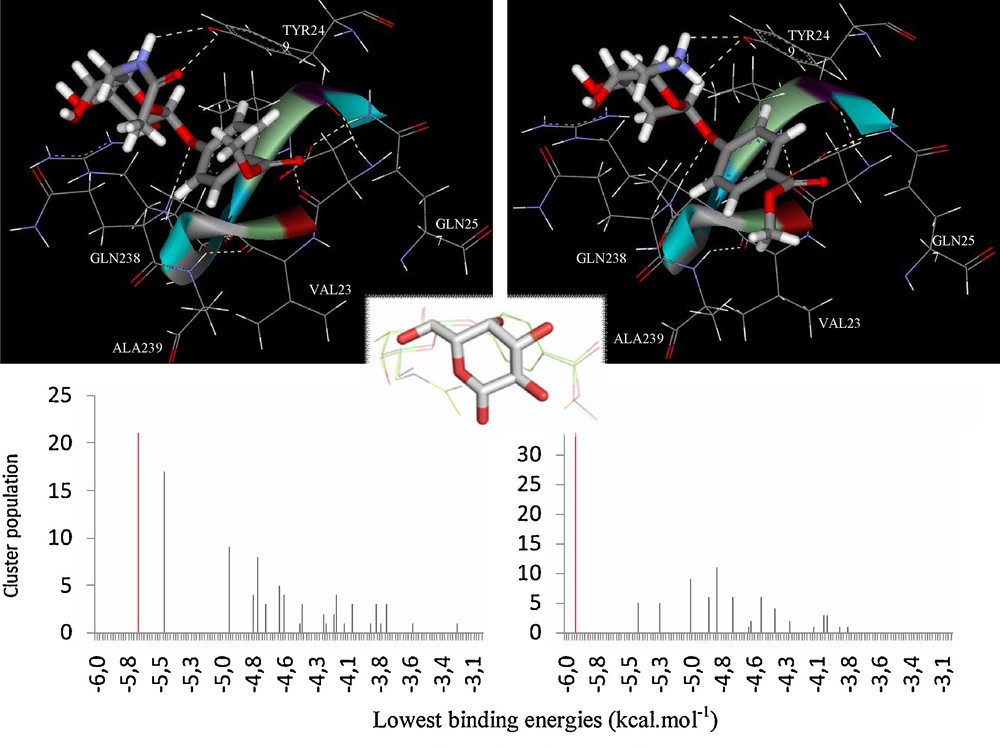
Docking results for compounds 7 and 8 complexed at the VAA Tyr249 site. The conformers representing the highest populated, lowest energy clusters are tridimensionally presented at the top; only the amino acids located inside an 8 Å radius (centered on the ligand) are displayed; hydrogen bonds are shown in dashed yellow lines. The docking histograms are shown below, where the mentioned conformers are highlighted in red. In the center, the overlap of β-D-galactose and 7 and 8 is shown, stating the slightly different positioning, although at the same site.
The replacement of the anomeric hydroxyl by different sulfamide groups proved to be unfavourable (or highly unfavourable!) modifications, providing firm evidence of the importance of the anomeric OH group in the recognition event.
3 Conclusion
A fast screening method based on competition STD-NMR experiments was applied to the toxic lectin VAA and a library of synthetic galactose derivatives. The STD competitive inhibition results show the two-way effect a substitution in the C6 position can have, with the incorporation of an amide or acetamide group shown to be the most favourable. This increased affinity was rationalized through docking calculations revealing the existence of newly formed hydrogen bonds with the protein. The reported results are of significant importance in the interactive process of design of toxin inhibitors, whose potency could be further increased when presented as glycoclusters. [38].
4 Experimental section
4.1 NMR experiments
All NMR experiments were carried out on a Bruker Avance DRX 500 MHz NMR spectrometer equipped with a 5 mm inverse probe head. Data was collected at 298 K using New Era 5 mm borosilicate tubes (reference NE-SL5-7). STD samples with VAA were prepared at a 60 μM protein and approximate 4 mM ligand concentrations (67:1 ligand to protein ratio), in a 99.9% deuterated PBS buffer ([NaCl] = 138 mM; [KCl] = 2.7 mM; [Na2HPO4] = 10.1 mM; [NaH2PO4] = 1.8 mM) at pD 7.5. STD-NMR experiments were recorded using the standard STD pulse sequence [39] with a 15 ms 5 MHz spin lock to minimize the background protein resonances. Saturation of the protein signal was performed using a train of 40 selective 70 dB Gaussian pulses of 50 ms duration, totalling a 2 s saturation time. The on-resonance frequency was set up at −1 ppm (VAA), and off-resonance was applied to 100 ppm. STD spectra were acquired with a total of 400 transients in addition to 16 scans to allow the system to return to equilibrium.
4.2 Docking simulations
The XR structure of VAA was extracted from the Protein Data Bank, PDB 1PUM. Ligands molecules were removed, hydrogens added, and bonds orders corrected using the Protein Preparation Wizard workflow implemented in Maestro 9.0 (Schrödinger, Inc.). Ligands were generated and minimized using the OPLS 2005 force field, in Maestro (Macromodel 9.0). A rigid receptor docking method was applied, where the ligand was treated as flexible and the protein held rigid. Receptor and ligand input files were generated in AutoDock Tools (AutoDock's 4.0 user's interface). Gasteiger charges were added to the ligand, and Kollman charges and solvatation parameters added to the protein. Search parameters were set to 100 runs for each complex, 25 000 000 evaluations per run, and a population size of 200. The mutation and crossover rates were set to 0.08 and 2.0, respectively, and the maximum number of generations was 27 000. A cubic grid box was defined with 20.7 Å side and centred in Tyr249 aromatic ring.