

1 Introduction
Carbohydrates at the cell surface (glycolipids and glycoproteins) play key roles in the recognition mechanisms of various biological cells and their external environment. Specific, reversible interactions between these carbohydrates and protein receptors (lectins) are of great importance in many normal and pathological biological and biochemical processes ranging from fertilization and intercellular communication to bacterial invasion and tumor metastasis [1]. The lectins typically exhibit high specificity and weak affinities toward their carbohydrate ligand. In nature, this low affinity is compensated by the architecture of the lectin itself, containing typically two or more carbohydrate binding sites, and by the host presenting the oligosaccharide ligands multivalently or as clusters on the cell surface [2]. The resulting interaction thus being a combination of several simultaneous binding events, the overall binding is significantly greater than the simple combination of the individual binding events, i.e. the “whole” of the interaction is greater than the sum of its parts. This effect is known generally as multivalence or the cluster-glycoside effect with reference to carbohydrate interactions, and is well documented for lectin–carbohydrate interactions [3,4].
Due to their function, lectins are important targets for many analytical, diagnostic or therapeutic applications. In order to study and potentially interfere, control or block these interactions, many multivalent “scaffolds” have been developed. Synthetic oligosaccharides, themselves functionalized with suitable “spacer” molecules, have been conjugated to a number of multivalent scaffolds from carrier proteins [5], dendrimers [6–9], micelles [10] and polymers [11,12] to nanoparticles and quantum dots [13–15]. Several examples of gold nanoparticles functionalized with biologically active oligosaccharides (glyconanoparticles [GNPs]) can be found in the literature [16,17], as well as a myriad of other examples of nanoparticles coated with other biologically important molecules exhibiting a multivalent effect [15,18–21].
The aim of the present article is to review the thermodynamic models that are prevailing in the study of multivalent interactions, which characterize protein–carbohydrate interactions. The investigation of such multivalent interactions has generated the synthesis of a range of multivalent scaffolds, based on various sub-structures, as platforms for presenting carbohydrate ligands from monosaccharides to larger oligosaccharides. These synthetic and semi-synthetic scaffolds are reviewed along with a critical appraisal of their functionalities on the molecular basis, which underlines the expression of the cluster glycoside effect.
2 Multivalence
The valence of a material, an atom or molecule, macromolecule, protein or indeed a cell, is defined as the number of separate, identical connections that can be made between that material and its binding partner(s). In general, this can be defined as a material presenting n binding sites which bind to n ligands, where n ≥ 1. This, of course, would include the case where a material presenting n binding sites interacts with a second material presenting n ligands, thus forming n interactions, where both the binding sites and ligands are presented multivalently (Fig. 1).
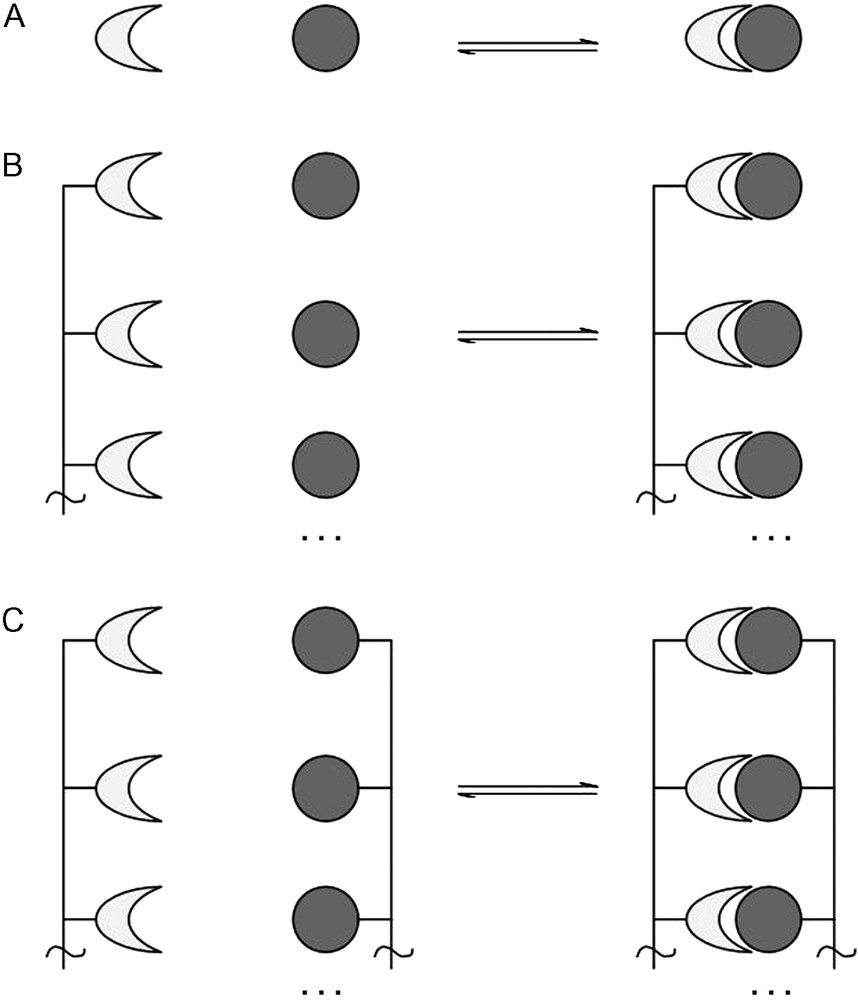
A. Monovalent receptor binding with a monovalent ligand. B. Receptors presented multivalently binding with several monovalent ligands. C. Receptors presented multivalently binding with ligands presented multivalently.
These multivalent interactions occur frequently in a wide range of situations in biological systems as well as in the synthetic world. For example, the pairing of deoxyribonucleic acids may be considered as multivalent; as one DNA monomer pairs with its complementary partner, hydrogen bonds form between the two molecules. As a molecular pair, these hydrogen bonds are easily broken, resulting in the starting monomers. However, when a single strand of DNA binds to its complementary strand, all of the bases align with their binding partners to form the double helix and a multitude of hydrogen bonds are formed. The intermolecular hydrogen bonding between the two strands becomes significantly more stable, so stable that enzymes, high-energy radiation or chemical modification are required to break the DNA double strand.
Therefore multivalence incorporates another dimension to binding interactions as the physical, chemical and biological surfaces may indeed change upon the occurrence of multiple binding events. Indeed, at the biological level, many molecules which form non-covalent bonds exhibit only weak binding affinities for their ligands (compared to, say, biotin-streptavidin Kd = 1 × 10−15 M, Ka = 1015 M−1). If several binding domains are presented multivalently, the overall binding affinity of its ligands increases. This would result in a change in the physical properties of the non-covalent bonds formed, making the global interaction much more important.
The multivalent behavior exhibited by carbohydrate–lectin interactions is particularly interesting in this respect. Carbohydrates are found in large quantities and in a multitude of structural variations conjugated to proteins, peptides and lipids, giving the glycoproteins, glycopeptides and glycolipids, respectively. These glycoconjugates can be found presented at biological interfaces including 2D surfaces and 3D clusters. Due to the diverse structural arrangements of such molecules, an enormous amount of information ranging from cell-type, development stage and pathological status can be stored within their structure [22,23]. Given the quantity of information that can be written using glycoconjugates, and the many ways of presenting this information, reading this glycocode is a complex process carried out by carbohydrate recognition proteins, the lectins. Like their carbohydrate ligands, the presentation of lectin binding domains can be equally complex, having to present several binding domains in order to simultaneously capture several ligands.
2.1 Types of multivalency
Multivalence can range from one molecule to clusters, self-assembled monolayers and 3D platforms. Biological systems use various types of multivalency depending on the local environment and/or a particular function. In particular, lectins exhibit diverse architectures and modes of multivalence relating to their function. Some typical examples can be found in Fig. 2. Con A [24] (in pH neutral environments) and PA-IL [25], for example, are both tetrameric proteins with tetrahedral and planar structures respectively, with all carbohydrate binding sites facing different directions. PA-IL and BclA [26] are soluble lectins implied in bacterial invasion and adhesion [27], whose roles may be to explore local environments (in solution or on surfaces), therefore their structures may reflect this purpose (Fig. 2). The cholera toxin has what is known as an AB5 architecture with the lectin domains forming a pentameric structure, with the receptor binding sites located on the same face of the macromolecule, and all oriented in the same direction [28]. This is indicative of its function – finding a suitable invasion site at the host epithelial surface [29].
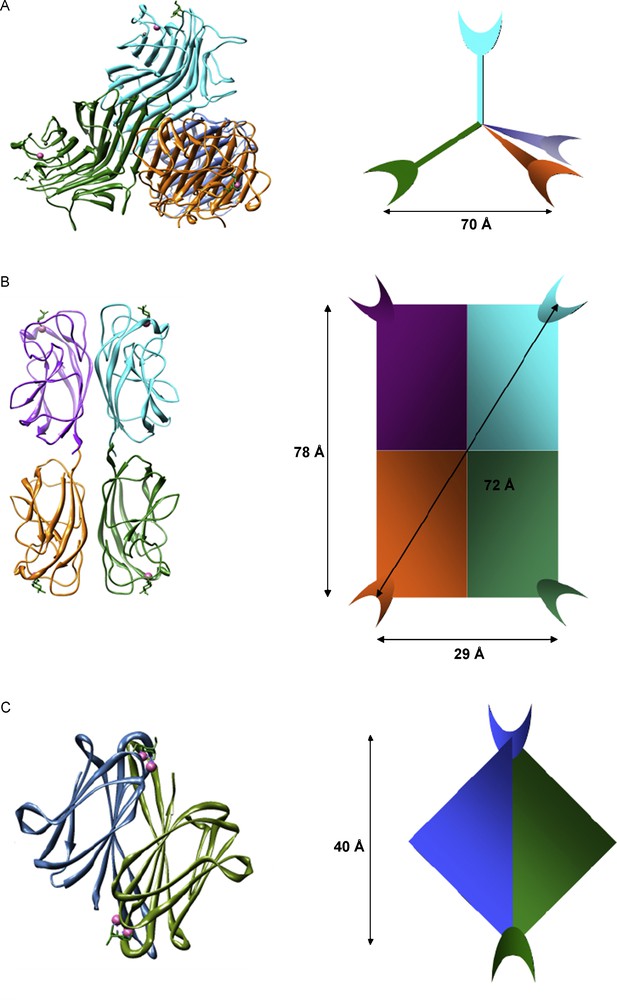
A. Crystal structure of Con A complexed with Me-α-d-Man and its tetrameric architecture (note 1). B. Crystal structure of PA-IL complexed with Me-β-d-Gal and its square planar architecture (note 2). C. Crystal structure of BclA complexed with Me-α-d-Man and its dimeric architecture (note 3).
These lectins exhibit “architectural multivalence”, consisting of one macromolecular structure which presents several, equivalent carbohydrate recognition domains. Other lectins, such as the adhesins, are membrane bound and consist of a lectin structure which presents only one carbohydrate recognition domain, attached to fimbriae or flagella tethered to the cell surface [30,31]. Several fimbrial structures clustered together at the cell surface would also give a multivalent presentation of the lectins in the extracellular environment. The galectins, which are membrane bound and specific for β-galactosides, as well as other C-type lectins, exhibit many different macrostructural formations giving rise to several different forms of multivalent presentation. Galectins-1 and 2 form homodimers in solution (like BclA), whereas Galectin-3 exhibits a carbohydrate recognition (lectin) domain tethered to a non-lectin domain which forms oligomeric structures in solution and on surfaces. Other galectins (galectin-4, galectin-8) form tandem repeat units, where the two carbohydrate recognition sub-units are attached via a (relatively) flexible linking molecule [30,32]. The collectins are a group of mannose binding lectins (C-type III lectins) that associate together forming oligomers. Three sub-units combine to form a trimer; the trimers are bound together via a collagen-like α-helix which is tethered to the cell membrane. Trimer structures associate to form larger oligomers such as bouquets of trimers (where all binding domains are presented adjacent to one another) or cruciform (cross-like) structures where four bouquet trimer sets present their recognition domains in four different directions [30,32,33]. Biological systems take advantage of several modes of multivalence in many normal and pathological processes depending on their position, function and status (Fig. 3). Often, these systems will present a wide variety of different “tools” in order to increase the likelihood of successful interactions or invasion pathways.
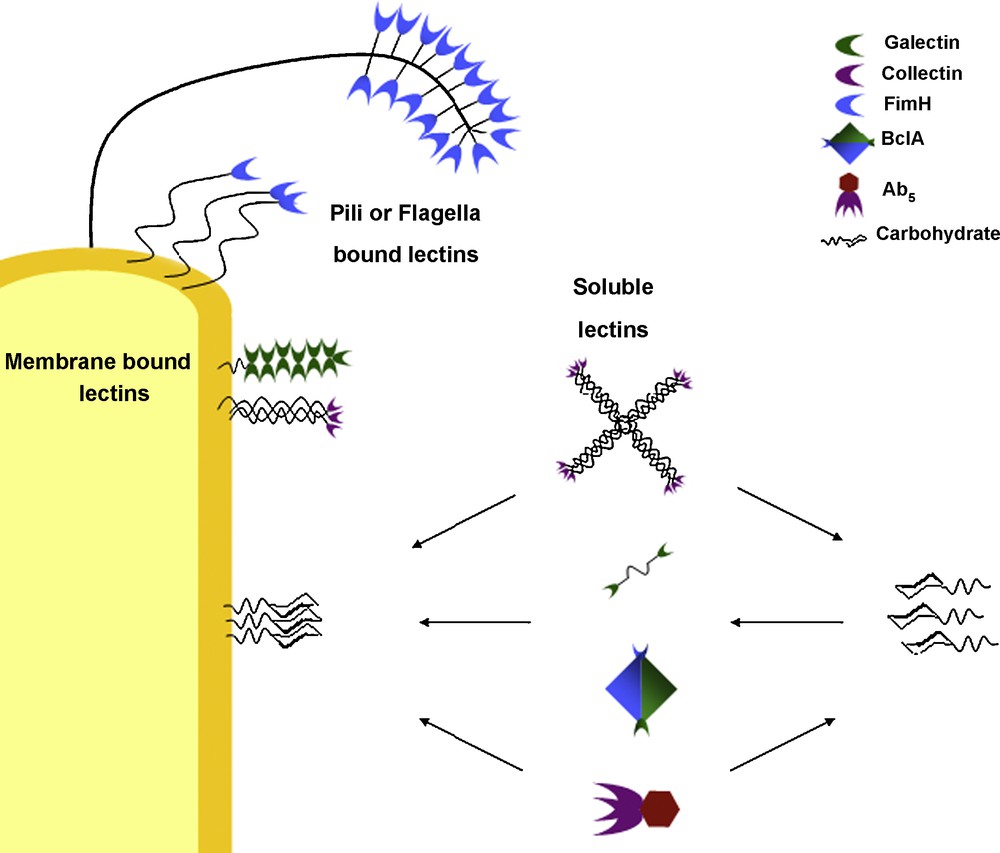
Different modes of multivalence observed in biological systems (not to scale).
3 Cluster glycoside effect
Increasing the clustering of receptors allows for several identical interactions to take place. This increase in local valency will therefore increase the overall interaction strength. This is often seen in biological systems in order to increase the interaction strength of typically weak binding partners and has been suggested as being evolutionary in nature, being easier to multiply the number of binding sites than developing more complex binding partners [4]. However, an effect which is seen throughout biology where multivalence occurs is that the global interaction appears stronger than the sum of the individual monovalent interactions. This phenomenon is often seen between lectins and their carbohydrate binding partners, and is known as the “cluster glycoside effect” [34]. Typically, the cluster glycoside effect is common to most lectins. However, this effect is noted even more so when the multivalent lectins bind with carbohydrates that are also presented multivalently. It is thought that a complementarity is observed between the binding sites of the lectin and the carbohydrate molecules which augments binding strength.
In Fig. 4A, one can see a single glycolipid molecule consisting of a Sialyl Lewis X oligosaccharide conjugated to a Ceramide chain. Several of these molecules have then been fixed to a 2D surface, as they would be presented at the cell surface with the lipidic portion forming part of the cell membrane and the carbohydrate portion extending into the extracellular matrix. The diagrams Figs. 4B and C show the top view of this glycolipid surface, as seen by an extracellular object such as a lectin. The diagram in Fig. 4C shows the molecules colored by hydrogen-bonding potential. Clearly, the interactions experienced by the lectin at the outside of the cell is not only a multiplication of several monovalent interactions, rather it experiences the effects of a binding surface of several closely-knit molecules. One analogy is the presentation of a single tree, when in fact one should consider the whole forest canopy. In the following paragraphs, the principles and the kinetic and thermodynamic explanations of the cluster glycoside effect will be explained with reference to lectin–carbohydrate interactions.
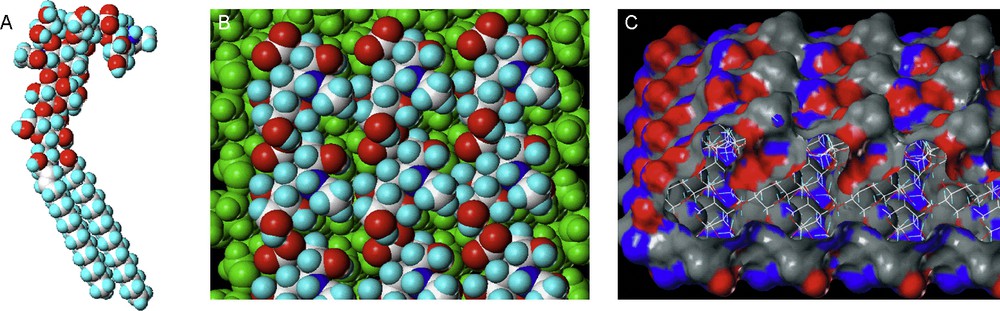
SialylLeX-ceramide conjugate (A). Several SialylLeX conjugates fixed to a 2D surface, view from above, coloured by atom type (B) and hydrogen bond potential (C).
The most important review to discuss the multivalent effect in recent years is that of Mammen et al. [2]. This review covers in detail the thermodynamics and kinetics of multivalent interactions in general; in particular they discuss cooperativity and introduce the affinity enhancement coefficients as well as a comprehensive survey of polyvalency in biological systems. Here, we have developed these themes further, with a particular emphasis on lectin–carbohydrate interactions.
3.1 Monovalent reaction kinetics
First, interactions of a ligand to its receptor at the monovalent level shall be considered (Fig. 2A). We will assume that the reaction follows the typical reaction kinetics and thermodynamics where:
A is one binding partner, B is the second binding partner, AB is the bound complex and k represents the rate constant for the forward (association rate constant, ka, M−1 s−1) and reverse (dissociation rate constant, kd, s−1) reactions. Equilibrium association constants (Ka, M−1) and equilibrium dissociation rate constants (Kd, M) can be calculated from ka and kd from the equation:
From the van’t Hoff equation, one can link the effects of rate constants (kinetic parameters) to the Gibbs free energy (G, a thermodynamic parameter).
The Gibbs free energy is in turn associated with the enthalpy (H) and entropy (S) of the interaction. From this, the dependence of kinetics and thermodynamics have over each other can be seen, as well as their dependence on temperature.
3.2 Monovalent thermodynamics
3.2.1 Enthalpy
The change in enthalpy, ΔH, is the difference between the final and initial enthalpic states of all components in the reaction, and can be separated into several components:
ΔHAB represents the enthalpy change upon bond formation between the two reactants. In the case of lectin–carbohydrate interactions, these non-covalent bonds may be hydrogen bonds between the hydroxyl groups of the carbohydrate and the amino acids of the binding pocket, as well as coordination bonds to metal ions or indeed aromatic stacking and stabilization of hydrophobic surfaces on either binding partner. Van der Waals and dipole–dipole interactions may also be included in this term.
ΔHconf represents the enthalpy change required to induce a particular conformational change of either binding partner. For example, the rotation of hydroxyl groups in order to maximize hydrogen bond formation, the rotation of glycosidic bonds or the twisting or displacement of any large amino acid groups to better accommodate the ligand such as the histidine residue around the binding pocket of BclA [26], or the “tyrosine gate” in FimH [35].
ΔHsol is the solvent contribution to the enthalpy of the interaction. More specifically, this would represent the making and breaking of hydrogen bonds between the solvent molecules and the binding pocket, the carbohydrate ligands and bulk solvent upon the displacement of ordered water molecules during lectin–carbohydrate binding (Fig. 5).
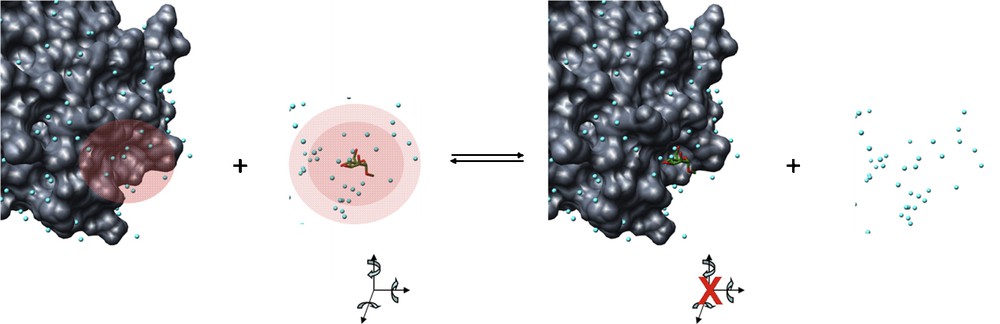
Enthalpy changes during lectin–carbohydrate interactions. The red shaded regions represent regions of ordered water, where hydrogen bonds with solvent molecules would occur. Upon interaction, these hydrogen bonds are broken and replaced by non-covalent bonds between the lectin and the carbohydrate. These regions of ordered water are thus expelled to the bulk solvent where they form hydrogen bonds between themselves and the bulk solvent. Carbohydrate degrees of freedom (translation and rotation about x, y and z axes) as well as conformational freedom about the glycosidic bond and hydroxyl groups would be restricted in the protein binding pocket.
3.2.2 Entropy
The change in entropy, ΔS, is the difference between the final and initial entropic states of all components in the reaction and represents the change in disorder of the system during association of the reactants. Like enthalpy, entropy can be separated into several components:
The translational entropy, ΔStrans, of a molecule arises from its freedom to translate independently through space in any of the x, y and z axes. It also refers to the translation of individual atoms or groups of atoms within the molecule, giving rise to bending and stretching of covalent bonds within the molecule; however, these will not be considered here.
ΔSrot refers to the entropy of rotation which arises from the free rotation about the principal axes by the molecule. ΔSrot is also related to the rotational freedom of individual atoms and groups of atoms. The protein structure will not change greatly upon interaction with its ligand. However, particular groups in or around the binding pocket may find themselves restricted rotationally upon ligand binding where before they were not. However, the carbohydrate will experience conformational restrictions imposed by the sterics of the binding pocket, ΔSconf. In particular, the rotational and translational freedom of the carbohydrate molecule will be dramatically reduced upon entering the binding site. In free solution, the carbohydrate is free to rotate and translate in all (x, y and z) dimensions. As the carbohydrate becomes bound, translation will be restricted as the lectin–carbohydrate complex becomes one entity. Rotation about the axes will be severely restricted due to the multiple non-covalent bonds formed between the reactants. Hydroxyl groups involved in the binding event will be “fixed” by hydrogen bonding or coordination to metal ions, with the primary hydroxyl groups of C6 likely to experience these restrictions more so than the secondary hydroxyls of carbon atoms in the saccharide ring. Glycosidic torsions will also be restricted due to the steric requirements of the binding site which would lock the carbohydrate in a particular conformation.
Finally, ordered solvent molecules in the binding pocket and those surrounding the saccharide molecules will be displaced to the bulk solvent upon interaction, contributing to an increase in the disorder of the system, ΔSsol (Fig. 5).
3.3 Monovalent to multivalent
Described above are the various contributions to the enthalpy and entropy of a monovalent lectin–carbohydrate interaction. These in turn influence the Gibbs free energy of this monovalent event which itself influences the kinetics of the interaction. The association constant represents the affinity of the lectin for its monovalent ligand. However, on a multivalent scale, one must also consider additional contributions to the enthalpy and entropy and their influence on the kinetics of the interaction.
3.3.1 Multivalent kinetics and free energies
Fig. 1A presented the monovalent association of two binding partners. As the valency increases, the free energy released upon multiple independent interactions increases also. With a given number of n independent interactions, the quantity of free energy released can be denoted as ΔGmul,n. The average free energy released can be described as the total free energy released for all interactions divided by the total number, n, of interactions.
Similar statements can be made with respect to the association constant, Ka, using the van’t Hoff equation where:
Assuming of course that the interactions were independent of one another, one can see that in fact and .
However, multivalent lectin–carbohydrate interactions often exhibit a Ka much greater than that seen for their monovalent counterparts. Therefore, there are several factors that cannot be explained by the increase in valency. As these factors result in increased kinetic activity of the interaction, they must play a significant role in the thermodynamics of the system on a molecular level.
3.3.2 Cooperativity
The average free energy released upon the interaction of a lectin binding site presented multivalently and their ligands (which may or may not be presented multivalently) may be equal to its monovalent analogue. However, the average free energy released may also be greater than, or less than the monovalent analogue. This difference is defined as the “cooperativity”, the degree of which is defined as α.
If α is equal to one, no cooperativity is observed in the multivalent interaction in comparison to its monovalent analogue. Positive and negative cooperativity is observed if α is greater than or less than one, respectively. Cooperativity will thus have a direct effect on the kinetics of the interaction:
Positive cooperativity occurs when the interaction of one ligand to one binding site increases the affinity of the other binding sites, facilitating following binding events. Negative cooperativity would occur when the binding of the first ligand to the first binding site impedes the binding of the second ligand to the following site.
However, as the cooperativity factor has an exponential influence on Ka, an interaction which has an enhanced multivalent association with respect to the monomer may exhibit negative cooperativity but a significant affinity enhancement all the same, i.e. tight binding would not necessarily require positive cooperativity, only sufficient affinity enhancement (α > 1/n). This has been labeled the affinity enhancement factor, β, by Mammen et al. [2], where the enhancement factor is a ratio of the global equilibrium association constant, , to the monomeric equilibrium association constant :
In terms of free energies, this becomes:
When investigating the interaction between a multivalent receptor and several monovalent ligands, cooperativity and enhanced affinity typically emerge from structural changes in the receptor which are induced by the first binding event. From the perspective of the ligands, no change occurs – each individual ligand acts independently to the others, and does so without memory. However, when considering the binding of a multivalent ligand to a multivalent receptor, the binding of one ligand would direct or hinder the following binding events, depending on the nature of their multivalent presentation, which in turn would influence the thermodynamics and kinetics of the following interactions. In order to study the reason for this affinity enhancement, one must look closer at the enhanced free energies and their enthalpic and entropic contributions to explain what results in this enhanced affinity, cooperative or not, from presenting ligands in a multivalent fashion.
3.3.3 Enthalpies of multivalent interactions
The multimeric free energy, ΔGmul,n, consists of its enthalpic and entropic components where:
The case of a dimeric lectin interacting with two monovalent ligands is shown in Fig. 6A. The two monovalent ligands interact with the two binding sites in the same fashion. Therefore, the enthalpy involved in the dimeric interaction is double the amount of the monovalent interaction. In the case of a divalent ligand interacting with the divalent receptor, several situations can occur depending on the nature of the linkage between the ligands. Firstly, the linking molecule can be of ideal length to present the two ligands in a fashion complementary to the receptor binding sites (case B). This means that no effort is required by the linker to modify the presentation of its ligands, its enthalpic contribution is negligible and the second intramolecular interaction occurs as in case A. In case C, the linking molecule is shorter than the ideal length. The first interaction occurs as before, as this first interaction is independent of the linker molecule. However, for the second intramolecular interaction to occur, the receptor has to alter the position and orientation or conformation of the binding sites. This invokes an energy penalty, reducing the observed enthalpy for this second interaction. Likewise for situation D where the linker is too long. The second interaction has to pay a conformational enthalpy penalty in order for the linker molecules to present the ligands in the correct fashion. In this case, the flexibility of the linker will play an important role. A rigid linker would have to pay a large conformational enthalpy penalty, whereas a flexible linker would pay only a small penalty. Generally, the more conformationally rigid a multivalent entity is, the more likely small, spatial mismatches between ligands and receptors will result in enthalpically diminished interactions [2].
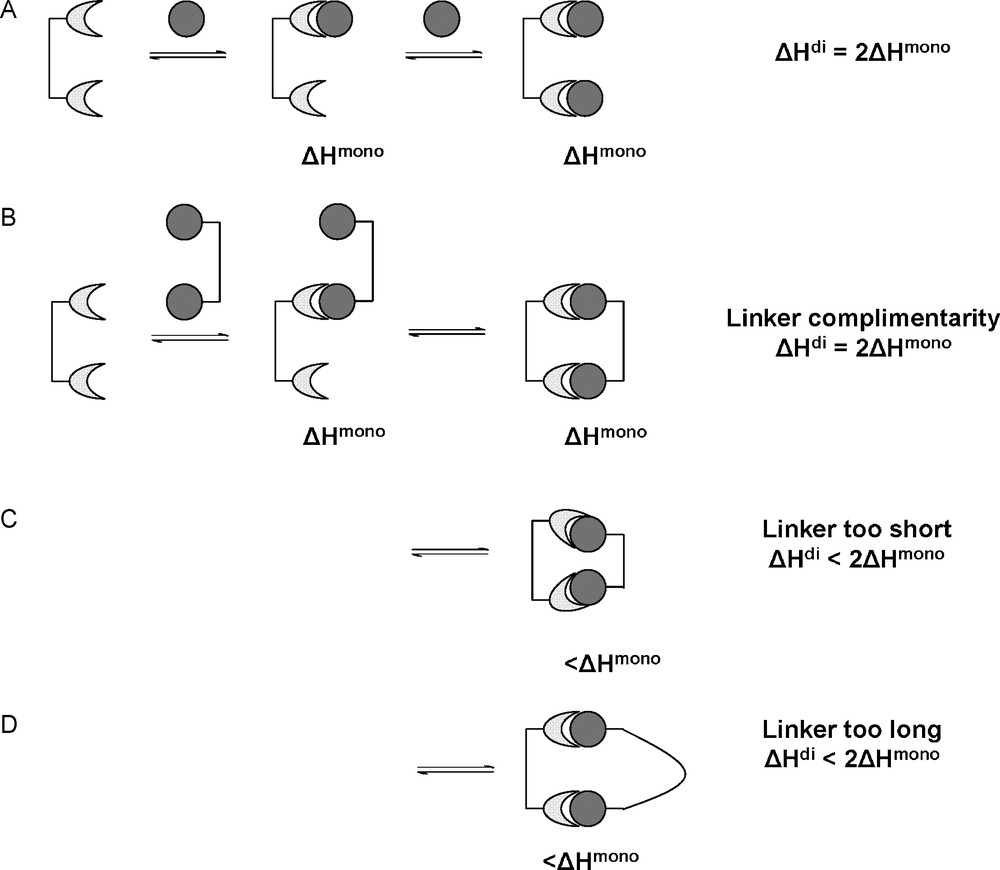
Bivalent receptor interacting with monovalent ligands (A) and bivalent ligands under ideal (B) and non-ideal circumstances (C,D).
In case C, where the linker molecule is too short, it is unlikely that the receptor will alter its conformation in order to accept the second ligand in an intramolecular interaction. Probably, the receptor will accept only one ligand presented, the second binding site being free to interact with a second bivalent ligand intermolecularly, thus removing the enthalpic penalty (Fig. 7A). In case D, where a long, rigid linker is used, it may be more beneficial for the bivalent linker to bind to two separate receptor molecules (Fig. 7B). The above situations where intermolecular association is more favorable than intramolecular association may lead to the formation of aggregates via ligand-receptor cross-linking. These aggregates would develop in length and become insoluble in the media resulting in irreversible precipitation.
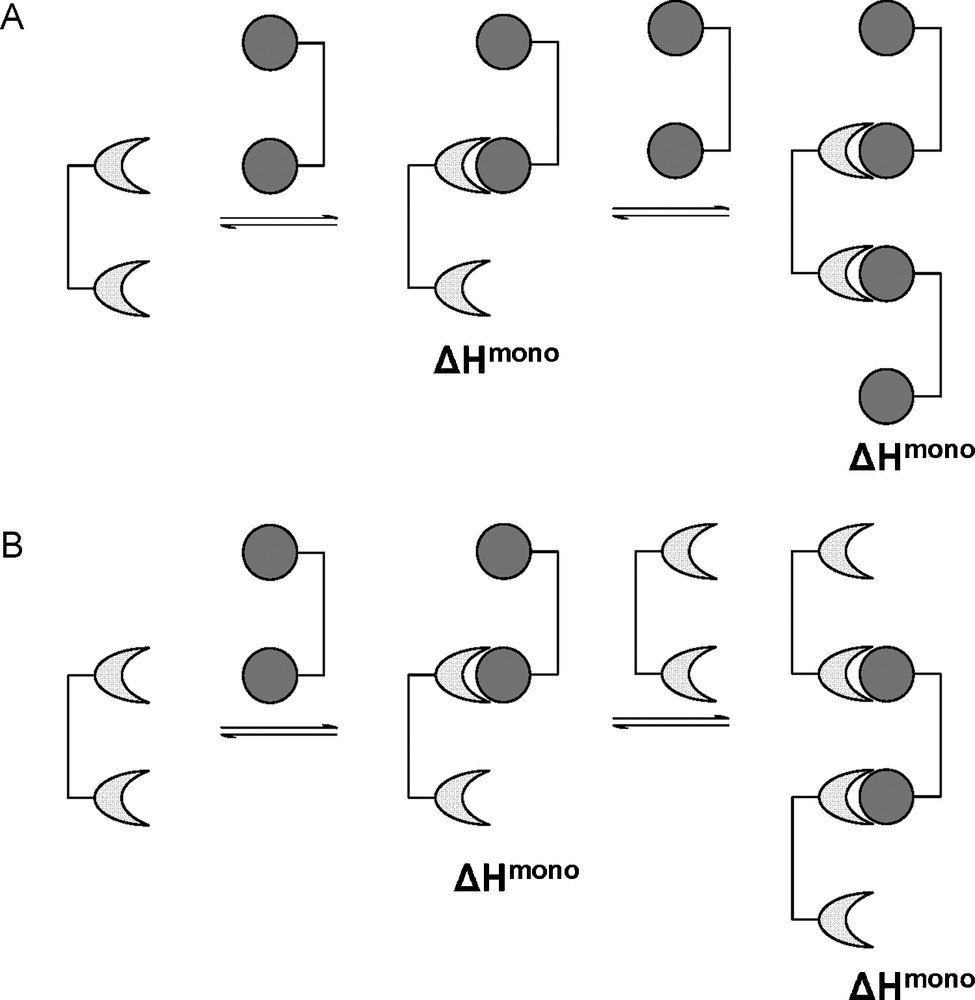
The alternative intermolecular interactions for bivalent ligands of non-ideal presentation.
One can summarize the multivalent enthalpy change as contributions from the monovalent enthalpy change and the conformational enthalpy dictated by the linker molecule used in the multivalent ligand architecture as follows:
3.3.4 Entropies of multivalent interactions
When moving from a monovalent to a multivalent system, the entropies involved change dramatically, particularly when considering intramolecular associations. As with the case for enthalpy, the multivalent entropy change (ΔSmul) will be related to the entropy change experienced by each ligand with respect to translational and rotational degrees of freedom of the ligand as it is immobilized in the binding pocket, as well as the release of ordered water molecules (ΔSmono). This can also be expressed in terms of cooperativity:
This would imply that a value of α greater than one would equate to an entropically enhanced multivalent interaction, whereas α less than one would imply an entropically diminished multivalent interactions.
However, in the multivalent system, one must also include the effect of linker molecule conformation (ΔSlink,conf) and solvent effects (ΔSlink,sol). Thus, the multivalent entropy contribution can be broken down further and described as:
In the monovalent system, the entropy change was related to the rotational and translational degrees of freedom of the ligand and receptors before and after intermolecular association as well as the entropy related to conformation and solvent effects. By considering case A presented in Fig. 8, the interaction of a bivalent receptor with two monovalent ligands, the change in entropy (ΔSmono) will be the same for both interactions as, again, the two ligands will be interacting independently of each other and without memory. In terms of molecularity, this would be disfavored entropically as it would involve the conversion of three entities into one, causing the loss of a total of 12 degrees of translational and rotational freedom (three translational and three rotational for each ligand). This loss of independent translational and rotational freedom has also been referred to as the Gibbs connections energy [36]. In case B (Fig. 8B), where the bivalent ligand is ideally presented, the interaction of a bivalent ligand would again be entropically disfavored (however less so than the monovalent situation) as this would involve the conversion of only two entities to one, causing the loss of only six degrees of rotational and translational freedom i.e. the entropic cost of the perfect bivalent ligand will be half of that of two monovalent ligands. It is for this reason that ΔSmono has to be corrected to for multivalent ligands:
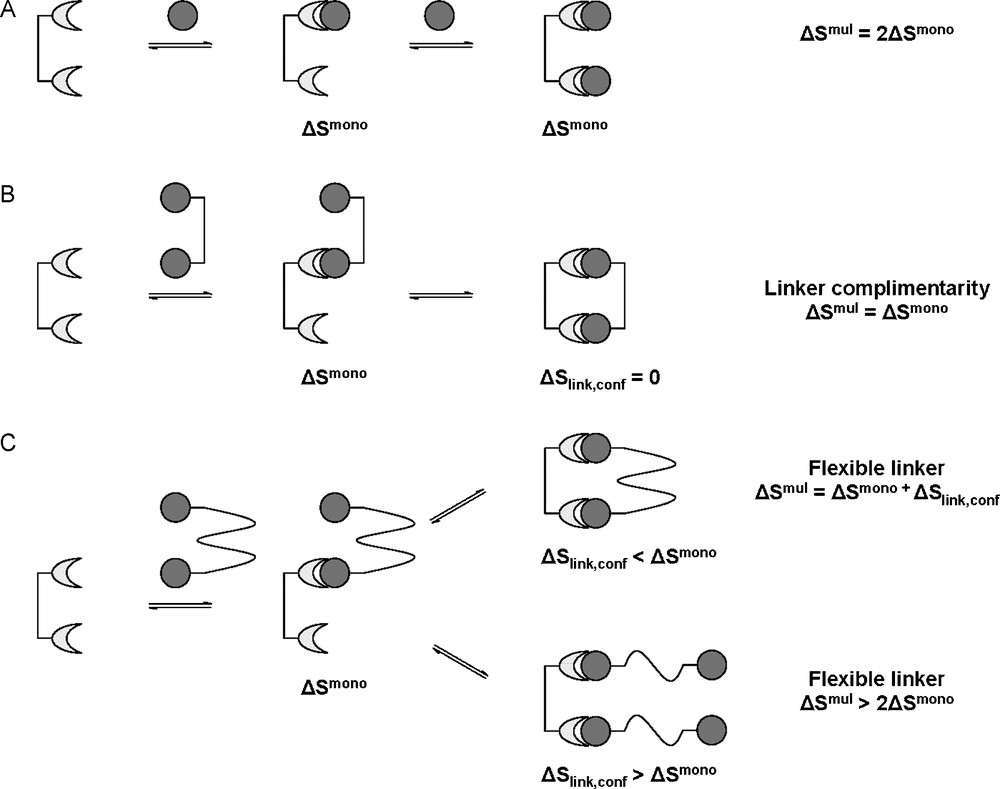
Change in entropy for multivalent interactions. Interaction with monovalent ligands (A). When the linker molecule is perfect for intramolecular association (B). When the linker molecule is too flexible (C).
The ΔSlink,conf term in the above statement comes from the inclusion of the linker molecule and its associated degrees of freedom. Assuming that the linker molecule is again perfectly shaped, rigid and conformationally locked (allows for no free rotation about its bonds), the entropy change for intramolecular association (ΔSlink,conf) will effectively be zero. This, however, is very unlikely, as all linker molecules would assume some form of flexibility (meaning ΔSlink,conf < 0, unfavorable), Fig. 8C. A linker molecule which is suitable and rigid would require only small entropic constraints (ΔSlink,conf < ΔSmono) and it would therefore be in the interests of both molecules to form the second intramolecular interaction, still being entropically enhanced. If, however, the linker molecule is unsuitably shaped or too flexible, this conformational entropy penalty would be very large. If this conformational entropy penalty is larger than the entropic penalty of the monomeric interaction ΔSlink,conf > ΔSmono, it would become more economical to form a second intermolecular interaction than to continue with intramolecular association, as the intramolecular interaction would no longer be entropically enhanced. In the case where the entropic penalty of linker conformation equals that of the monomeric interaction, an equilibrium between intra- and intermolecular interactions would occur.
ΔSlink,sol comes from the effect of the linker molecule on the local solvated environment. Assuming that the linker molecule does not affect the ligand-receptor interactions, ΔSlink,sol will be zero. However, this may not always be the case. The linker molecule may disrupt the organization of water molecules around the ligand before or during interaction. Therefore, the organized water around the ligand which would be displaced by the receptor binding site upon multivalent interaction may be different to the monovalent interaction due to the influence of the linker molecule. Likewise, organized water molecules around the linker molecule, which would not be present in the monovalent ligand, may find themselves displaced by the receptor binding site. Also, if the linker molecule interacts in some way (hydrophobic interactions or aromatic stacking for example), the linker may displace ordered water molecules associated with the protein surface which may not have otherwise been disturbed. Finally, the ligand in the monovalent form may organize water around itself in a particular fashion. Upon multivalent presentation, independent of the linker molecule, ligand molecules may associate together by forming close-knit networks of hydrogen bonds and ordered water molecules. Carbohydrate molecules are particularly effective at organizing intermolecular hydrogen bond networks (Fig. 9) [37].
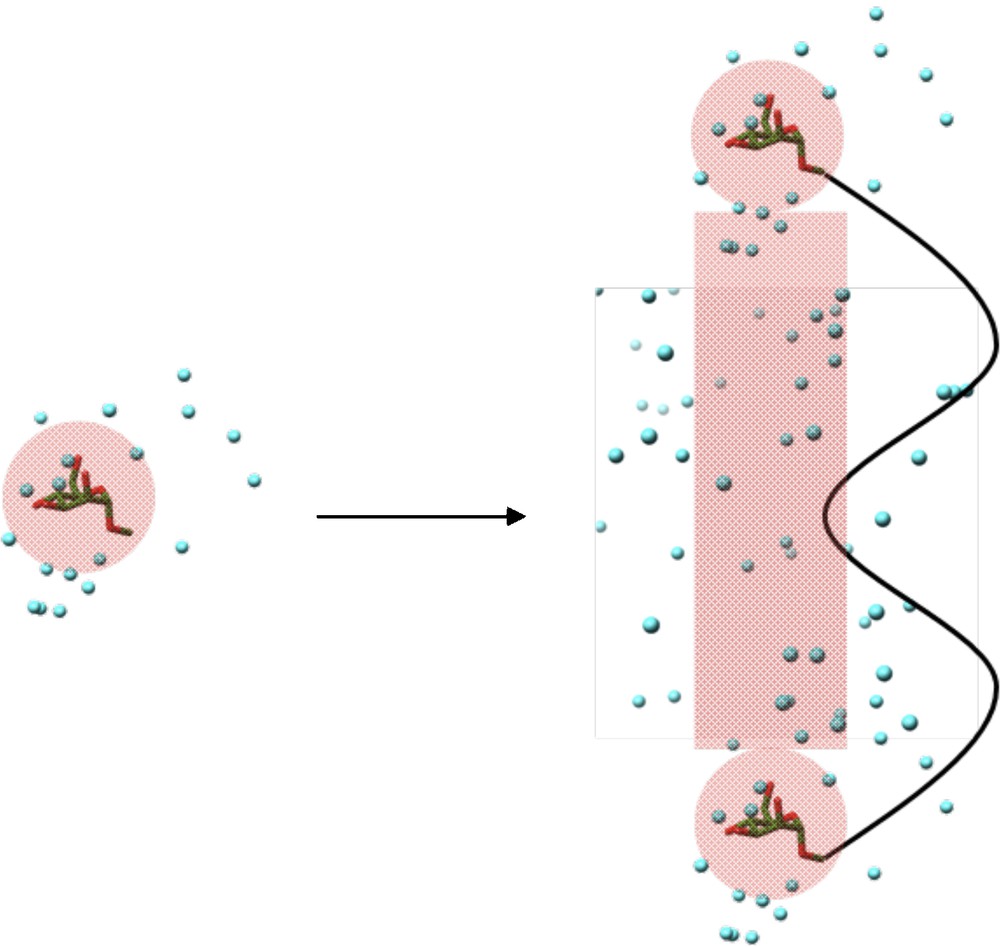
Representation of inter-ligand hydrogen bond networks formed upon multivalent ligand presentation. Regions of ordered water molecules shaded in red.
3.3.5 Enthalpic and entropic contributions to multivalent-free energies of interaction
Above we have discussed the enthalpic and entropic contributions to the Gibbs free energies for both monovalent and multivalent binding. Both the enthalpic and entropic contributions can influence whether an interaction will proceed intramolecularly and multivalently or intermolecularly. From the monovalent to multivalent situations, many factors must be considered as the global interaction becomes more complex. Clearly, the nature of the linker plays a crucial role in influencing ligand-receptor interactions at the multivalent level. In order to influence multivalent binding over aggregation, linker molecules need to be designed correctly with respect to the target receptor. Presentation of ligand molecules with the correct spacings and conformationally rigid architectures should be used in order to minimize the conformational restraints imposed on the ligands. Some degree of flexibility should also be incorporated to maximize the fitting of the ligand-receptor pairs. These are all important factors in multivalent binding from a thermodynamic point of view.
3.4 Effective concentration
The above description of the multivalent effect is based solely on the thermodynamics of the system, which in turn define the Gibbs free energy and its resultant effect on the equilibrium constants. Essentially, the multivalence effect (and thus the cluster glycoside effect) was analyzed in terms of entropy and is a concept brought by Mammen et al. [2]. An alternative binding model can be used to describe multivalence, in terms of effective concentrations (Ceff) which was proposed by Kramer and Karpen and applied to carbohydrate–protein interactions by Gargano et al. [38,39]. The example by Mulder et al. describes the interaction of a β-cyclodextrin receptor dimer with a bis-adamantyl ligand [40]. The bivalent interaction should again be treated as a two-stage system with an initial intermolecular interaction, followed by a second intramolecular interaction. This first interaction obliges the second ligand of the dimer to be in close proximity to the second available binding site. Therefore, this second binding site experiences a localized high concentration of neighboring free ligands. If this local concentration density is greater than the ligand concentration in the bulk solution, intramolecular (multivalent) binding will be favored [41]. Thus, the term Ceff represents this local concentration density experienced by both the ligand and receptor. The theory of effective concentration thus gives a concentration-dependent association constant for the second binding event. At low concentrations of ligand, the effective concentration experienced by a multivalent receptor due to a multivalent ligand would be much greater than the concentration of free ligands in the bulk solution (Ceff ≫ Cbulk) and thus a multimeric intramolecular interaction is more likely to occur. However, at higher ligand concentrations, (Ceff < Cbulk), the concentration of ligand experienced by the receptor will not be influenced by the ligand molecule already bound and therefore a second, intermolecular interaction is more likely to occur (Fig. 10). Naturally, when Ceff = Cbulk, the system will be in equilibrium between the intra- and intermolecular interactions.
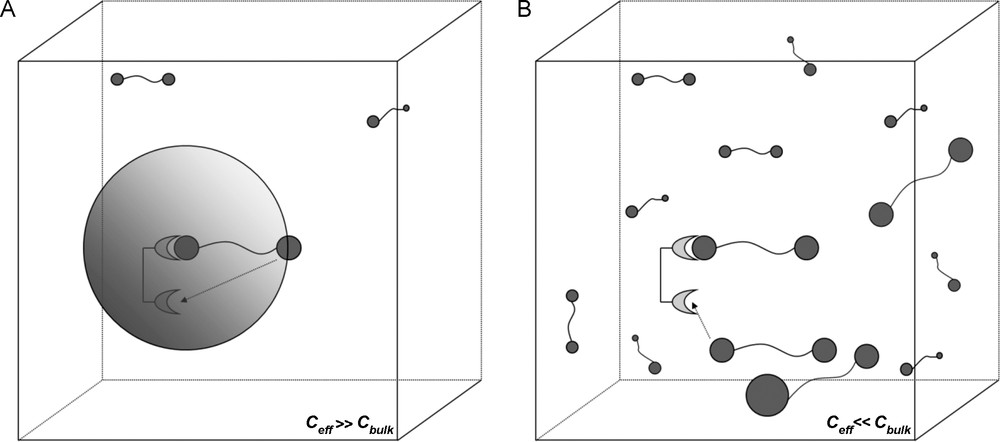
A. Concentration dependence of multivalent interactions. Intramolecular association more likely as Ceff ≫ Cbulk. (A). The shaded sphere represents the “probing volume” that the ligand can use to search for other binding sites. Intermolecular association more likely as Ceff < < Cbulk (B).
Ceff is dependent on the ligand-receptor distance, which is also dependent on the length of the linker molecule. Ceff will also be influenced by linker molecule flexibility. The length, of course, determines the radius of the sphere occupied by both potential interacting partners. Longer molecular lengths lead to smaller Ceff values, decreasing cubically with the increase in length. Therefore it is important that the linker molecule is long enough and flexible enough to allow the intramolecular interaction, but not too long so that Ceff approaches Cbulk. The flexibility of the linker molecule would also influence the nature of the theoretical sphere occupied by the second ligand after the first binding event has occurred. A flexible ligand would theoretically be allowed to occupy any position within this sphere (excluding regions occupied by the receptor or the other parts of the ligand). A less flexible linker molecule would reduce the sphere in size or dimensionality depending on the nature of its rigid properties. In this reduced system, the Ceff would be increased, increasing the probability of the intramolecular interaction occurring. Too rigid, however, and the intramolecular interaction may be inhibited thermodynamically (enthalpy or entropy penalties) or logistically (the second ligand is simply blocked from entering the second binding site).
3.4.1 Reaction kinetics and the effective concentration
For a divalent receptor interacting with a divalent ligand in the example given by Mulder et al., the first binding event would occur more readily by a factor of four in comparison to its monomeric counterpart [40], due solely to statistical considerations. These statistic considerations are the ratio of possible permutations of the forward reaction, leading to the monomerically bound complex, and the permutations of the reverse reaction, dissociation back to the reactants (Fig. 11) [39].
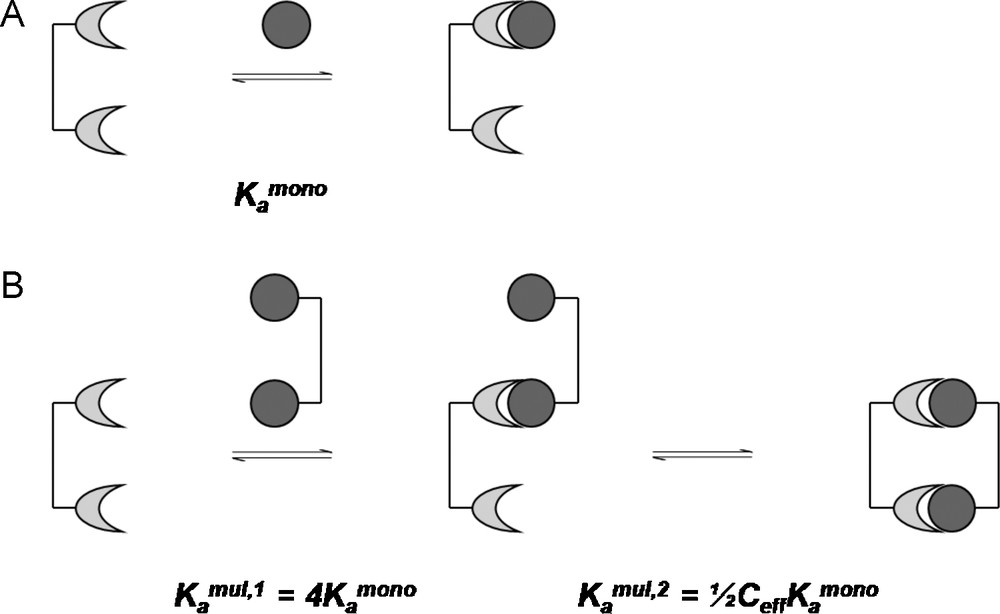
Kinetics of a divalent interaction by considering the effective concentration.
The overall binding constant for the divalent interaction being dependent on Ceff then becomes:
A theoretical estimate of Ceff (mM) is cited as:
If we consider the case presented in Fig. 11 again, the monomerically bound complex is an intermediate between the reactants in free solution and the multivalently bound complex. This intermediate could be regarded as being in a ‘steady state’. The lifetime of this intermediate would then depend on (the dissociation rate constant from the intermediate back to free reactants) and (the association rate constant from the intermediate to the multivalently bound complex). If the association/dissociation rate constants (i.e. and ) are low in magnitude, the lifetime of this intermediate species may be sufficient for the effective concentration to have an effect on the conversion to the multivalent species. If however is large and monovalent dissociation a very fast process, the lifetime of the intermediate would be very short, meaning that there would be a higher probability the ligand would escape the proximity of the binding sites. Similarly, if was large in magnitude, the intermediate species would be too short-lived as to experience any beneficial effects offered by the effective concentration, being swiftly and independently converted to the multivalently-bound complex.
4 Synthetic multivalent scaffolds
For the study of multivalent interactions, in particular lectin–carbohydrate interactions, many research groups have synthesized a range of multivalent scaffolds based on various sub-structures as platforms for presenting carbohydrate ligands, from monosaccharides to larger oligosaccharides. Natural, synthetic and semi-synthetic scaffolds used range from small-molecule clusters, to dendrimers, polymers and micelles, as well as 2D surfaces for applications to inhibit or elicit a biological response [3]. Scaffolds can vary in size, valency, what form they adopt in solution and their physical properties. Different scaffolds allow different topologies which leads to a particular presentation of the carbohydrate epitopes.
Different scaffolds with the same valence may exhibit different levels of activity towards the same lectin receptor. Likewise, the same multivalent structure may exhibit different affinity trends towards one lectin than to another, as shown by Brewer's group for interactions of Con A and Dioclea grandiflora lectin (DGL) [43]. As discussed above, the scaffold, acting as the linker molecule between ligand epitopes, plays an important role in ligand activity and its mechanism of action. The architecture of the scaffold may also be tailored to influence the macromolecular assembly upon ligand-receptor association. The scaffold used may also be tuned to a particular type of multivalence or to a particular location – binding to a multimeric lectin such as Con A may require a different multivalent ligand to that binding to several fimbriae-tethered lectins such as FimH. Likewise, a membrane bound lectin would prefer a multivalent scaffold different to that of a soluble lectin – a 2D SAM as opposed to a 3D structure, for example. Therefore, by manipulating the multivalent scaffold, one can be selective to the target receptor, as well as incorporate other functionalities (e.g. fluorescence). The valence of the multivalent structure is also important. Often, increasing the valency of the structure will increase the affinity of the lectin, to a point where further increasing the valency no longer has any, or only very little, effect on the Ka of the interaction [6]. Also, the valence of the structure may influence the formation of the macromolecular products. Sisu et al. showed that increasing the valency altered the kinetics and aggregation mechanisms resulting in intermolecular cross-linked aggregates as opposed to intramolecular interactions [44]. It may also be the case that, where a particular structure was an effective inhibitor in monomeric form, when grafted to a multivalent scaffold, the inhibitor may lose some potency. This is because one must consider inter-ligand interactions which would not occur at the monovalent level, but may dramatically affect ligand presentation on a multivalent scaffold.
4.1 Small molecule glycoclusters
A great number and variety of ligands have been reported with the specific purpose of investigating multivalent interactions. A large proportion of these include small clusters (dimers, trimers, tetramers, etc.) fixed on to small multivalent scaffolds of varying size and flexibilities. Often, these small synthetic molecules can incorporate multiple functionalities (Fig. 12A).
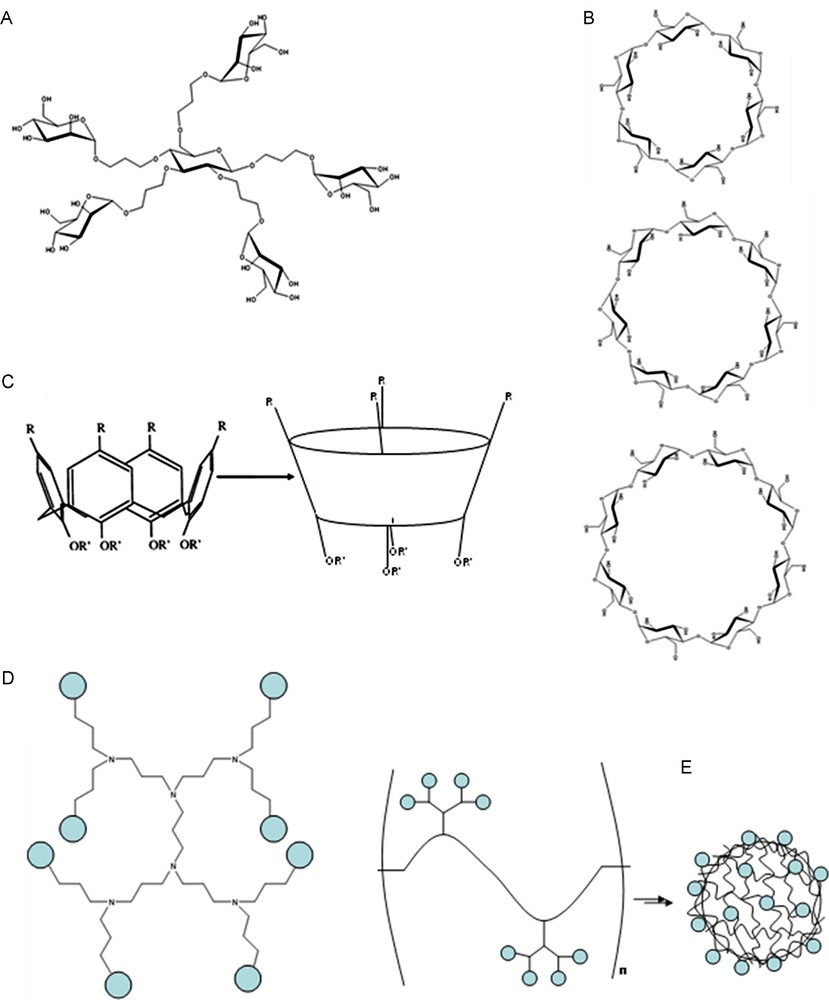
A. Example of one of several Mannose functionalized “octopus” glycoclusters developed by Dubber et al. for the inhibition of FimH [51]. B. Structures of α-, β-, and γ-cyclodextrins (top, middle and bottom respectively). C. Structure of calix[4]arenes. D. Third generation poly(propylene imine) dendritic backbone. E. Schematic representation of functionalized polymer structures aggregating to form capsules.
Brewer's group tested several small divalent glycoclusters when investigating interactions with Con A and DGL [43,45]. Several of these clusters were developed around aromatic (alkyne or benzylic) scaffolds, giving very short, rigid structures. Also presented are dimers connected by alkyl chains of varying length or aromatic rings bound via thiourea bridges, again of varying length. The same group investigated di-, tri- and tetravalent trimannoside structures with a longer, more flexible thiourea bridge to an aromatic core [45]. All multivalent structures showed some improvement in Ka with enhancements ranging from ∼2- to 10-fold for the monosaccharide structures and up to 1000-fold for the tri-mannosides in the case of Con A. More impressive improvements were observed for DGL. Summaries can be found in Table 1 for the most efficient of each cluster type. Isothermal titration microcalorimetry (ITC) was used to determine the Ka of each cluster. Toone's group also investigated the cluster glycoside effect observed in Con A [46]. Several multivalent clusters were developed around an aromatic core allowing the synthesis of di-, tri-, tetra- and hexamannosides, the most potent of which being the dimer structure, shown in Table 1. It must be noted that the results from Toone's group are presented with respect to mannose equivalents [46], whereas results from Brewer's group [43,45] are presented with respect to the whole cluster.
Summary of various multivalent scaffolds found in the literature.
| Multivalent Platform | Carbohydrate motif | Target lectin | Ka M−1 | Relative Affinity Score | n | Measured by | Reference |
| Me-α-d-Man | Me-α-d-Man | Con A | 1.20E + 04 | 1a | 1 | ITC | [43] |
| Alkyne Linker | α-d-Man | Con A | 2.50E + 04 | 2.1a | 0.52 | ITC | [43] |
| Alkyl Linker | α-d-Man | Con A | 5.30E + 04 | 4.4a | 0.54 | ITC | [43] |
| Aromatic-thiourea | α-d-Man | Con A | 4.50E + 04 | 5.7a | 0.6 | ITC | [43] |
| Aromatic-thiourea | 3,6-di-O-(α-d-Man)-α-d-Man | Con A | 6.50E + 07 | 1170a | 0.26 | ITC | [45] |
| Aromatic | α-d-Man | Con A | 2.4 | 1 | ITC | [46] | |
| 6th Generation PAMAM dendrimer | α-d-Man | Con A | 350 | Haemagglutination | [68] | ||
| 6th Generation PAMAM dendrimer | α-d-Man | Con A | 5350 | Haemagglutination | [72] | ||
| Me-α-D-Man | Me-α-d-Man | DGL | 4.60E + 03 | 1 | 1 | ITC | [43] |
| Alkyne Linker | α-d-Man | DGL | 2.20E + 04 | 4.8 | 0.68 | ITC | [43] |
| Alkyl Linker | α-d-Man | DGL | 1.06E + 05 | 23 | 0.56 | ITC | [43] |
| Aromatic-thiourea | α-d-Man | DGL | 4.50E + 04 | 9.8 | 0.52 | ITC | [43] |
| Aromatic-thioureaa | 3,6-di-O-(α-d-Man)-α-d-Man | DGL | 6.50E + 07 | 14,000 | 0.25 | ITC | [45] |
| β-Galactoside | Gal | PA-IL | 6.7E + 3 | 1 | 1 | ITC | [66] |
| 2:2 Calixarene | Gal | PA-IL | 5.7E + 6 | 852 | 4 | ITC | [66] |
| Lewis a | Lewis a | PA-IIL | 4.70E + 06 | 1 | 1.08 | ITC | [47] |
| bis-triazole functioalized tetraethylene glycol | Lewis a | PA-IIL | 1.10E + 07 | 2.3 | 0.66 | ITC | [47] |
| tris-triazole functionalized aromatic ring | Lewis a | PA-IIL | 9.68E + 06 | 2.1 | 1.11 | ITC | [47] |
| GlcNAc | GlcNAc | WGA | 1 | Haemagglutination | [58] | ||
| Cyclodextrin | GlcNAc | WGA | 280 | Haemagglutination | [58] | ||
| Gal | Gal | ECorL | 1 | Haemagglutination | [58] | ||
| Gal-CD | Gal | ECorL | 14b | Haemagglutination | [58] | ||
| Lactose | Lactose | ECorL | 1 | Haemagglutination | [58] | ||
| Cyclodextrin | Lactose | ECorL | 14b | Haemagglutination | [58] | ||
| LacNAc | LacNAc | ECorL | 1 | Haemagglutination | [58] | ||
| Cyclodextrin | LacNAc | ECorL | 28b | Haemagglutination | [58] | ||
| Lactose | Lactose | PNA | 1 | ELLA | [59] | ||
| Penta-cyclodextrin functionalized polymers | Lactose | PNA | 4 | ELLA | [59] | ||
| Pk | Pk | SLT-I | 1 | 1 | ELISA | [79] | |
| STARFISH | Pk | SLT-I | 106–107 | 5 | ELISA | [79] |
a Not per mannose, per molecule.
b Reactivity per heptamer, not per galactose/lactose.
Other small glycoclusters used for lectin–carbohydrate interactions were developed by Roy's group for the PA-IIL lectin [47]. These scaffolds include aromatic centers or ethylene glycol chains coupled to Lea moieties via triazole rings via the commonly used “click” chemistry. Several dimers and trimers were synthesized, the most potent of which are listed in Table 1.
Other glycoclusters include mannose functionalized pentaerythritol and bis-pentaerythritol scaffolds developed by Roy's group for the inhibition of Con A activity on Mesenchymal stromal cells and the inhibition of the FimH lectin [48,49]. As well as aromatic molecules, other scaffolds can be found in the literature. Lindhorst's group used carbohydrates as the scaffold base, with mannose functionalized linkers coupled to the pendant hydroxyl groups (Fig. 12A) [50–52]. These “octopus” clusters were used to study multivalence and inhibiting Con A and FimH. They also added further functionalities to several of these clusters [53].
In relation to the application of the cluster glycoside effect, small molecules have proven to be very efficient. The di-, tri-, tetra- and hexavalent scaffolds designed by the groups of Brewer [43,45] and Toone [46] all show increased affinities towards their target ligand. However, in considering the thermodynamic and kinetic applications of the cluster glycoside effect with respect to the interactions studied, the inter-binding site distance of the lectin is significantly larger than the inter-ligand distance. This would lead to the situation described in Fig. 6C, where it is more likely that the lectin will choose to pay the entropic penalty and interact with two separate ligands (intermolecular association) than pay the enthalpy penalty of internal structural change for intramolecular association. This, of course, means that the ligands designed exhibit an effective valency of one (when considering one multivalent ligand interacting with one lectin). However, Dam et al. [43,45] observed that n values calculated from calorimetry data were inversely proportional to the functional valency of the multivalent ligand. This demonstrates that all carbohydrate epitopes were still available and interacting with a lectin binding site. This leads to the conclusion, which they also show with microscopy data, that cross-linking occurs, where a network of lectin–ligand complexes are formed, driven by intermolecular association. They also show that multivalent scaffolds with flexible linker molecules exhibit greater changes in enthalpy but larger entropy penalties as predicted earlier – flexible linkers allow optimum ligand-receptor interactions, yet a larger entropy penalty has to be paid resulting from constricted movement of flexible groups. This still leads, however, to an enthalpically enhanced affinity. This was also observed by Dimick et al. with the aggregation of tetrameric Con A [46]. Enthalpically enhanced affinities which are partly compensated by unfavorable entropic penalties. However, in both cases, enthalpic enhancement may not be the only cause of increased affinity. Due to their small sizes, these multivalent molecules exhibit a high local concentration of ligands. Therefore, upon lectin–ligand association, there may be a greater chance of a successful binding event with one of several ligands presented on the scaffold. This, of course, is related to the effective ligand concentration and its statistical contribution, the multivalent scaffold providing a “high density” binding partner with more possible binding permutations. However, as experiments were not carried out with monomeric analogues, it is not possible to compare observed affinity enhancements with those calculated from differences in free energy changes.
The small molecules designed by Marotte et al. also exhibited interesting binding kinetics to PA-IIL [47]. As expected, the monomer equivalents exhibited normal monomeric kinetics to multimeric receptors. The dimers, based on both aromatic and polyethylene glycol scaffolds, showed that each carbohydrate epitope binds to one binding site. The observed enthalpy was shown to be double that of the monomer as both carbohydrate epitopes are available for binding. However, the entropy contribution to the system's thermodynamics is not proportional to the valency of the scaffold. In fact, a large entropy penalty is incurred which significantly reduces their binding affinities. Even though the aromatic and linear dimer structures are long enough to theoretically induce multiple binding events on the same lectin molecule, this was shown not to occur. One explanation for this is related to the orientation of the ligands, which in linear form point in opposite directions. Therefore, a large conformational change may be required to present the ligands in the correct manner to induce multivalent binding. The trimer exhibited a stoicheometry of 1, indicating that one trimer binds to only one binding site. Thermodynamically, trimer binding is more or less equivalent to monomer binding with reduced enthalpic enhancement and favorable entropic contributions due to its rigid nature.
Dubber et al., although not dealing with a multimeric lectin, showed the increased inhibitory power of their ligands via ELISA tests [51]. Even though no quantitative thermodynamic or kinetic data can be extracted, it is clear from the data that ligand presentations varied with the carbohydrate scaffold base. This, in turn, altered the binding/inhibiting properties of the ligands towards the bacterial lectins.
4.2 Cyclodextrins and calixarenes
Cyclodextrins are cyclic oligosaccharide molecules of units of α-(1-4)-linked glucose. They exist in three conformations: α-, β- and γ-cyclodextrin which contain six, seven and eight glucose units respectively (Fig. 12B). The structures present a hydrophobic cavity and a hydrophilic exterior and it is for this reason they are very popular in supramolecular chemistry and host–guest interactions; the cavity acts as a host for a wide range of guest molecules, as well as biocompatibility [54]. Reinhoudt's group used cyclodextrins and guest molecules as a model system for multivalence interactions [41,55]. The hydroxyl groups on the two faces of the cyclodextrin can be modified in order to present molecules multivalently. Several cyclodextrins have been functionalized with carbohydrates giving rise to various glycoclusters for use as drug delivery vehicles [56,57], as well as high affinity ligands for lectins. Furuike et al. modified β-cyclodextrins with galactose and lactose to study their enhanced binding with the WGA and ECorL lectins [58], showing an enhancement factor of 40 (per epitope) for GalNAc functionalized CDs with WGA and factors of 2 and 4 for Galactose/GalNAc and LacNAc – CDs respectively with ECorL. Lactose functionalized α-cyclodextrins have also been used for the inhibition of peanut agglutinin (PNA) [59].
Calix[n]arenes are similar to cyclodextrins in that they are cyclic ring structures with a hydrophobic cavity capable of hosting a small hydrophobic guest molecule. They are composed of cyclic phenol-formaldehyde oligomers (Fig. 12C), the number of rings in the structure denoted by the [n]. As with the cyclodextrins, n can be varied in order to tune the size of the hydrophobic cavity. The phenol functionality and aromatic nuclei can be converted to most functional groups to attach ligating units. Their conformation can also be altered (conical or tetrahedral), allowing tunability in the presentation mode of functional groups added to the scaffold [42,60] (and references therein). There are several examples of the addition of carbohydrate molecules to form glycoclusters [60,61]. Fujimoto et al. functionalized calix[4]arenes with galactose and glucosides to study their specificities with PNA and Con A respectively, with a view to using the calixarenes as site-specific drug delivery vectors [62]. Ungaro's group have synthesized several calixarenes ranging from n = 4 to n = 8 [63]. These calixarenes were functionalized with two to eight galactose or lactose moieties via thiourea bridges. A range of conformers was possible due to the flexibility of the large aromatic scaffold. Inhibition tests were carried out using Viscum album agglutinin (VAA), a galactose binding AB type plant toxin from Viscul album, and galectins -1, -3 and -4. Inhibitory enhancements of factors of 8, 1.5, 1 (no affinity enhancement) and 0.7 (diminished affinity) were observed. Affinity investigations with cholera toxin were also carried out by fluorescence spectroscopy and SPR, showing an affinity enhancement factor of 5 and 18, respectively, per sugar epitope emphasizing that different analysis methods may give different results [64]. Recent studies using galactose functionalized calixarenes tethered to a microarray for studying the binding of RCA 120 and PA-IL lectins showed an affinity enhancement of 23 per sugar epitope for RCA 120 (no affinities recorded for PA-IL), as measured by fluorescence spectroscopy [65]. The results support the idea that different glycoclusters are required for different multivalent receptors.
For the interactions of ligands attached multivalently to cyclodextrin and calixarenes, there are few studies published providing fully quantitative information with regards to the thermodynamics of binding to multivalent lectins. This makes the thermodynamic interpretation of their multivalent properties difficult. Several studies have used ELISA and ELLA type assays to study their inhibition powers. Chwalek et al. showed that polymers functionalized with oligosaccharide functionalized cyclodextrins exhibited stronger inhibition powers per oligosaccharide compared to the monomer unit [59]. However, no significant synergic effects were noted, suggesting that there is no, or only very little, thermodynamic enhancement towards affinity augmentation. Again, this augmentation may be due to an increase in effective ligand densities. The studies of Ungaro's group were also conducted using inhibition effects [63]. They showed that the inhibition power of calixarenes varied between several lectins. This demonstrates that the calixarenes exhibit a relatively rigid platform which can be used for structure selective lectin targeting. This rigid nature would contribute to the increase in entropic enhancement to interaction affinity. A more recent study by Cecioni et al. uses calix[4]arenes functionalized with galactosides for inhibition/interaction studies of PA-IL binding [66]. Several topologies were developed in order to study the influence of calixarene structure and ligand presentation on lectin recognition. ITC studies revealed that a 2:2-trans conformation proved to be the most efficient binding partner, with an 850-fold increase in Ka. Results showed Gibbs free energies that were enthalpically enhanced in this conformation, yet with greater entropic penalties as multivalency increases, but nevertheless resulting in a net improvement of ΔG. Molecular modeling (docking) studies showed the potential for this particular topology to induce chelate binding to two of the binding sites on a PA-IL tetramer, which may lead to further aggregate structures. It is this chelation-aggregation which has been attributed to the multivalence effect observed with this particular scaffold.
There are of course many other cyclic structures that have been used for presenting carbohydrates for lectin recognition. Wilczewski et al. investigated multivalence and clustering using mannose regioselectively addressable functionalized templates (RAFT) polymers – cyclodecapeptide templates with up to four possible ligand attachment sites [67]. A second, regioselectively distinct site, can be used for adding other functionalities (biomarkers, functional groups for surface attachment, etc.).
4.3 Dendrons and dendrimers
Often, the small glycoclusters, such as those mentioned above, can be functionalized in such a way that they allow multimerization of the cluster groups themselves. This leads to higher order structures known as dendrons and dendrimers. Dendrimers have a regular branching pattern with predictable physical properties and display multiple copies of functional groups in a spherical arrangement [6]. Dendrons are branched structures with typically non-spherical arrangements; segments of whole dendrimers. Both dendrimers and dendrons have the capability of producing nanometer scale formations of very high valency (up to 400). The number and nature of the tethered functional groups can be controlled by the number of “generations” allowed, as can various physical properties such as solubility and reactivity [68]. The use of different sub-units would also allow for multiple functionalities. Several commercially available dendrimer frameworks include poly(amidoamine) (PAMAM), poly(propylene imine) (PPI, Fig. 12D) and poly(ethylene imine) (PEI) [6,69]. Several dendrimers with the PAMAM framework were developed by Cloninger's group and tested for their ability to bind with Con A, pea lectins and cyanovirin, stating an affinity enhancement of 350 and 0.38 (reduced activity) for Con A and pea lectins respectively [68,70,71]. Bifunctional dendrimers were also developed to alter mannose presentation and valency [72].
Roy's group built several dendron and dendrimer structures in order to inhibit Pseudomonas aeruginosa lectins to great effect [73,74]. The dendrimers developed included spherical and linear structures with a poly(lysine) and phosphodiester frameworks. Dual functionality (galactose and fucose) was incorporated in order to bind both PA-IL and PA-IIL. Other dendrimers were developed to inhibit the FimH lectin and as mimics of T-antigen markers from breast cancer, bearing a β-d-Gal-(1-3)-α-d-GalNAc moiety [7,75,76].
Trehalose-centred PAMAM dendrimers were developed by Lindhorst's group, following on from the octopus glycosides [50]. The pendant hydroxyl groups of the trehalose dimer were functionalized with the PAMAM framework, to which mannosides were attached via thiourea bridges. Several carbohydrate-based dendrons were developed for FimH inhibition [9,77]. There are many other examples of the use of carbohydrate functionalized dendrimers and dendrons in the literature using different core structures for studying different interactions [69,78].
One significant example is that of Bundle's group and the development of their STARFISH ligand [79]. This ligand is comprised of a glucose centre with each of the hydroxyl groups functionalized with divalent dendrons presenting Pk trisaccharides, giving rise to (up to and including) decavalent structures. The STARFISH was designed to inhibit shiga-like toxin (SLT) lectin systems which present carbohydrate recognition domains in a pentameric architecture such as that described earlier for cholera toxin. The presentation of the ligands on the starfish scaffold allowed complementarity towards the arrangement of the receptors, with each ligand occupying a binding site. Furthermore, ligand presentation not only allowed pentavalent binding to one SLT unit, but in fact two SLT binding units, with each arm of every dendron binding intermolecularly to two different SLT units. This resulted in an impressive 106–107-fold increase in ligand activity as seen by ELISA, with further improvements anticipated upon increasing the rigidity of the dendron arms.
There is little data available for quantitative measurements of thermodynamic contributions to the multivalence effect offered by dendrimers. ITC experiments by Mangold and Cloninger [70] showed that interactions with monomeric Con A demonstrated that all epitopes were available for binding. Increasing dendrimer valency lead to increased enthalpy contributions per epitope, but also increased entropy penalties, thus only marginally increasing the free energy of the interactions and lectin affinities. Interactions with dimeric Con A showed similar traits with respect to enthalpy and entropy, yet the affinity increases were much more exaggerated, suggesting that effective concentrations of both ligands and binding sites are important. However, in some cases, more than half of the carbohydrate epitopes were unavailable for binding. Typically, the larger generation dendrimers have larger binding constants, which could be a reflection on the larger inter-ligand distance and flexibility allowing for optimized interactions.
4.4 Functionalized polymers
Polymers are linear structures which present functional groups as branches from the main chain. Polymers, like dendrimers, are constructed by the polymerization of building units of natural or synthetic origin, and can be of controlled size and valency. Due to their potential large size, they can exhibit very high valency. Their nature and physical properties can be controlled by the use of different building blocks and side-chain functionalities resulting in highly tunable platforms. A wide range of building block molecules can be used to synthesize the polymer. Poly(phenylacetylene) was used by Otsuka et al. for the construction of glucose and galactose polymers for interaction studies with Con A and peanut agglutinin (PNG) lectins [80]. The polymers also gave chiral helical structures, the chirality imparted by the carbohydrate, which showed enhanced affinity towards the lectins. Block copolymers had also been developed by Lin et al. for the glucose sensitive aggregation of Con A [81]. Gestwicki et al. synthesized galactose functionalized polymers based on ROMP in order to investigate bacterial signaling in Escherichia coli [14]. Ogata et al. developed N-linked glycopolypeptides to inhibit influenza infections [82]. More recently, Zinger-Yosovich and Gilboa-Garber have used naturally occurring branched polysaccharides, normally used as food additives, for their anti-infection properties towards P. aeruginosa, Chromobacterium violaceum and Ralstonia solanacearum [83]. A further contribution from Bundle's research group includes the methodology of covalently pre-organized ligands presented on a polymer scaffold, designated as polyBAIT, which allowed for ligand activity enhancement of several orders of magnitude as seen by ELISA [84]. Natural polymers such as polypeptides can also be used as multivalent scaffolds [85].
As for several cases above, there is little quantitative data available for interpreting the thermodynamic and kinetic contributions of the cluster glycoside effect with polymer scaffolds. Dam et al. have conducted a thorough analysis of lectin binding to carbohydrate functionalized polymers [86]. They show that a “bind and slide”, or “bind and hop” model explains the large augmentations in lectin affinity. This model was designed to describe how enzymes bind to DNA, initially binding to a low affinity site then sliding along the DNA chain until a high affinity site is reached. In lectin–carbohydrate systems, the first lectin molecule binds with high affinity to the first available epitope. Dissociation–re-association occurs rapidly so that the overall kinetics show highly favorable binding. Upon saturation of the carbohydrate epitopes, subsequent binding (and re-binding) events occur with increasingly negative cooperativity. This resembles the effective concentration model for multivalent binding, with statistical effects strongly influencing the first and subsequent binding events – a reduction in further binding permutations upon saturation of the ligands presented on the polymer chain. The effective concentration model may not be able to fully explain this large augmentation in activity as steric effects may arise under high saturation conditions, reducing the affinity further.
4.5 Micelles and capsules
Molecules which consist of hydrophobic and hydrophilic building blocks allow for the construction of amphiphilic structures. When placed in polar solvents, the hydrophobic components collapse on themselves in order to be shielded from the aqueous environment. Simultaneously, the hydrophilic components expose themselves to the external environment, forming stable particles (Fig. 12E). This is one of the bases of the formation of capsules and micelles. Capsules are typically made of diblock polymers, whereas micelles consist of molecules which exhibit hydrophilic and hydrophobic components. If the hydrophilic components are functionalized with carbohydrates, these molecules can be presented on the micelle surface, in a multivalent arrangement. Micelles and capsules typically have a size range of nm – μm. Rieger et al. have produced several capsules as models for smart drug carriers, functionalized with mannosides, and tested with the BclA lectin [87,88]. Amphiphilic capsules based on polysaccharides (hyaluronic acid) and other biopolymers were also developed [89]. These hyaluronic acid capsules were developed by the layer by layer (LbL) adsorption of oppositely charged polymers around a sacrificial nanoparticle core. Iwasaki et al. also developed capsules to specifically target and tag non-natural carbohydrate molecules presented on carcinoma cells [90]. Micelles of mannose functionalized glycolipids have also been synthesized as multivalent ligands for Con A [91,92]. These nano- and microstructures present interesting candidates for intelligent drug delivery vectors as various biologically important molecules (drugs, fluorophores, etc.) can be trapped inside this hollow core to be released when and where necessary.
ITC studies conducted by Rieger et al. on polyethylene oxide – polycaprolactone micelles revealed that most of the carbohydrate epitopes were available for binding with the BclA lectin [88]. However, the enthalpy change of the interaction is slightly smaller, as is the entropy contribution, leading to a Ka one order of magnitude lower than that of Me-α-d-Mannose. This demonstrates a negative cluster glycoside effect, with no cooperativity or affinity enhancement.
4.6 Neoglycoproteins
Proteins and peptides are often glycosylated in nature, with glycosylation representing up to 70% of post-translational modifications [93]. Amino acids presenting O (serine or threonine) and N (asparagine) atoms on the peptide side chain provide potential glycosylation sites. Up to 90% of these naturally occurring glycosylation sites are occupied [23]. To produce this synthetically, however, solid state peptide methodologies have to be employed, requiring protection/deprotection of both carbohydrate and amino acids, and is limited to only 50 amino acids [93]. However, a variety of methods are available for introducing non-natural glycosylation sites. Typically, chemically modified carbohydrate molecules are coupled to modified amino acids in the peptide chain. Such examples include the use of thiohexoses which give thioether-linked (S-linked) glycoproteins upon coupling to sulfamidate-modified serine residues and Michael addition to unsaturated amino acids. Several site-specific methods have also been used such as disulfide bridge formation between thiohexoses (or functionalized thiohexoses: selenyl sulfides, methanethiosulfonates) and cystein residues [94].
Apart from only a few examples of their use as multivalent platforms for carbohydrate presentation, “glycodendriproteins” have so far not been widely used, despite their advantages as biocompatible, site-specific and targeting capabilities. Inhibition studies with galactose specific FimA and mannose specific Con A were reported; however, only a small (1.5-fold) increase in affinity enhancement was noted for Con A [95]. Yet a 20 nM inhibition of FimA was recorded, 106-enhancement compared to that of lactose [96]. One more prominent example in the use of proteins as multivalent scaffolds was developed by Bundle's group, following on from their STARFISH scaffold [97]. This elegant methodology employed an anti-nitrophenol antigen (anit-NP IgM) as a (up to and including) decavalent protein scaffold for presenting NeuAcα2-6Gal ligands to CD22 receptors. The organized assembly of antibody-CD22 complexes allowed for the assembly of antibody scaffolds of optimal inter-ligand distances for cell surface glycan binding.
The disadvantage of the use of neoglycoproteins is that they are often produced in heterogeneous mixtures, although the protein is coded for by DNA, the addition of carbohydrates is not controlled genetically. The carbohydrate functionalities are thus subject to various conditions such as enzymatic degradation, etc. which leads to the production of several different glycan structures, termed “glycoforms” [23,93].
As for, the functionalized polymers above, there is no information available describing the thermodynamic parameters of the cluster glycoside effect observed in neoglycoproteins. However, one can imagine that, even with modification of the amino acid sequence at particular positions, it would be very difficult to control the display and presentation of the carbohydrate ligands. It may also be difficult to control the valency and presentation density of these ligands, as increased conjugation of carbohydrate molecules may denature the protein, effectively giving an (oligo)saccharide functionalized polypeptide.
4.7 Self-assembled monolayers (SAMs)
Carbohydrate functionalized (glyco-)SAMs provide a 2D model of the glycocalyx. They allow control over ligand density and orientation in addition to being supported by analytical techniques such as microarrays, surface plasmon resonance (SPR), quartz crystal microbalances and atomic force microscopy (AFM). Several surfaces can be used to which saccharide molecules are mobilized. Typically, target molecules consist of a thio- or thioacetate functionalized linker molecule to allow the formation of a stable S-Au bond upon SAM formation, for which there are many examples [98–100]. The linker molecule will terminate in the carbohydrate epitope. Dilution of these carbohydrate functionalized alkane thiols with non-functionalized alkane thiols allows density control on the 2D matrix. The ligands form highly organized, densely packed, oriented monolayers. Although density control is possible, distribution is not, meaning that the inter-carbohydrate distance may not be regular throughout the SAM. With SPR applications, gold surfaces and SAMs were popularized, as were various surface immobilization methods. Dextran can typically be found coated onto the gold sensor chips to which a wide variety of functional groups can be coupled (amines, thiols, carbonyls and streptavidin-biotin coupling as well as hydrophobic and bilayer attachment). There are numerous examples of the use of monolayers for the multivalent presentation of carbohydrate ligands and their interactions with lectins. In microarray technology, several hundreds of amine functionalized glycans have been immobilized to functionalized glass slides. Plastics and modified cellulose have also been used to immobilize carbohydrate molecules [101]. Glycan arrays are often, if not always, used to determine the preferences of lectins for particular mono- or oligosaccharide structures and motifs.
Due to the limited dimensionality of SAMs, there are currently no experimental methods for evaluating the thermodynamic contribution to the multivalence effect – only affinity measurements by SPR or similar experiments. Also, the current technology for immobilizing molecules to 2D surfaces does not yet allow for a great deal of control. For example, one can control well the quantity of material immobilized to form the SAM, however, the distribution of ligands on the surface would be at random, assuming experimental design has no effect on ligand deposition (flow direction, flow speed, surface size, etc.).
4.8 Gold nanoparticles
There are many examples in the literature of carbohydrate functionalized gold nanoparticles – (glyconanoparticles, GNPs) for investigating carbohydrate interactions. GNPs are similar in size to many common biomacromolecules and provide an ideal scaffold for presenting carbohydrate molecules in a globular polyvalent configuration, much like the glycocalyx (Fig. 13). Also, the physical properties of the AuNPs described above may be applied to the detection and evaluation of their interactions [18,102]. Penades’ group have developed several GNP systems as a 3D model of the glycocalyx for studying carbohydrate–protein interactions [16,103,104]. Lin et al. synthesized mannose functionalized GNPs for the labeling of FimH on type I pili [105], as well as demonstrating an increase in lectin affinity for the GNP scaffolded mannosides. The same group studied the interaction of their mannose GNPs with Con A by SPR showing a 100-fold increase in inhibitory potential with regards to Me-α-d-Mannose. Kamerling's group also developed glucose and mannose functionalized GNPs for studying the interaction of Con A by SPR [15], UV/vis aggregation and TEM, showing an increased affinity of Con A for the GNP scaffolded mannosides. Chien et al. also used SPR competition studies to investigate the interaction between shiga-like toxin and globotriose functionalized GNPs [17]. Relative inhibitory potencies up to 200,000-times higher were observed for these GNPs with reference to the monomer ligand.
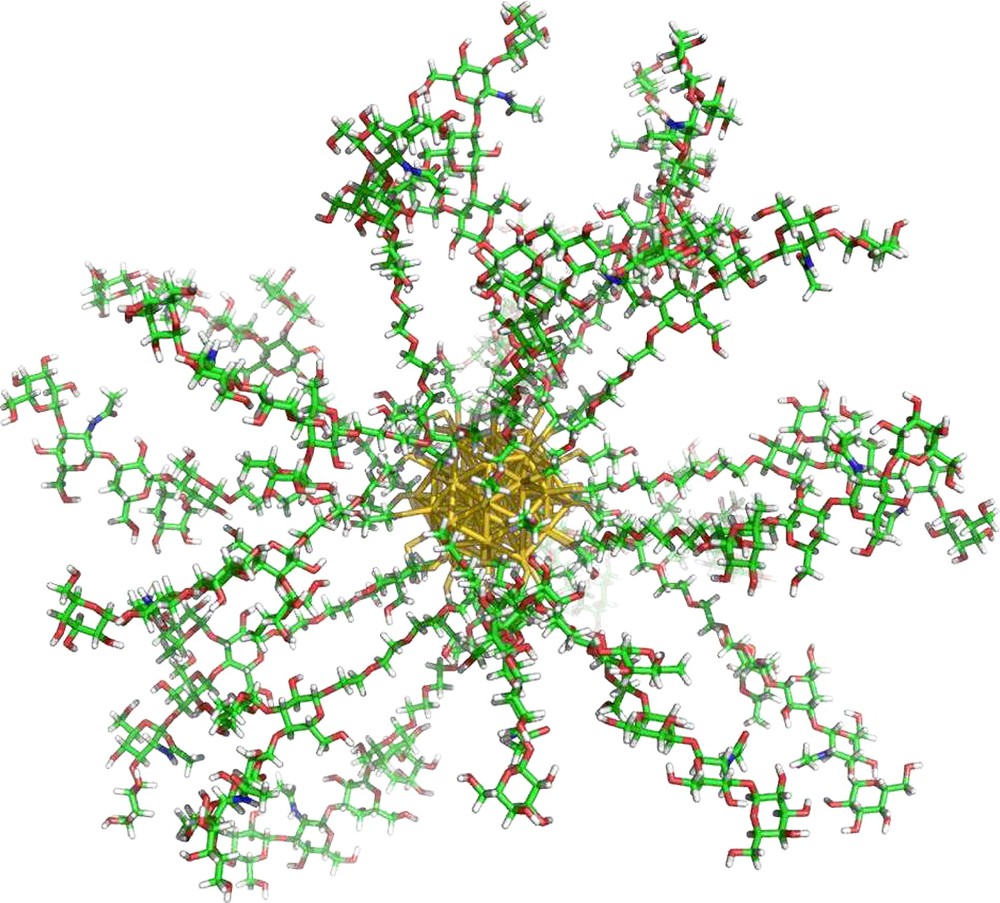
Gold glyconanoparticle.
The augmentation of lectin affinity for nanoparticle based carbohydrate ligands has been observed both qualitatively by haemagglutination inhibition assays with Con A and PA-IL as well as quantitatively using surface plasmon resonance, and the first use of isothermal titration microcalorimetry (ITC) for nanoparticle systems, for the BclA and PA-IL lectins [106]. As observed for BclA, hybrid GNPs with low density coverages (10, 25% presentation density in mannoside ligands) exhibited no improvement in lectin affinity in comparison with the ligand in free solution. This is thought to be because the inter-mannoside distance on the GNP surface is too great for the dimeric lectin to experience enthalpic complementarity, entropic enhancement or an increased local effective concentration to induce multivalent interactions. At 50% presentation density however, the Ka increases almost 40-fold, with enthalpic enhancement being strongly observed. Augmentation in PA-IL affinity was also observed for galactose functionalized GNPs by ITC. Ten-fold increases in interaction affinity were recorded from 17 to 33 and again to 100% galactose presentation densities. Although this augmentation was shown to be enthalpically enhanced, entropic enhancement also made a significant contribution. This confirms what was observed earlier by Barrientos et al. with the investigation of GNP presentation density on interactions with Viscum albumin agglutinin (lectin) and the enzyme E. coli β-galactosidase in competition and hydrolysis experiments respectively [16]. Investigations into the binding of mannose oligosaccharide functionalized GNPs with the DC-SIGN lectin also showed similar trends in HIV – DC-SIGN transfection and SPR inhibition studies [107,108].
In general, augmentation of Ka was observed when the inter-ligand distances approached half of those of the target lectin, suggesting that a combination of thermodynamic enhancement as well as effective concentrations and statistical considerations may be important for inducing multivalent interactions.
5 Conclusions
It was the aim of the present review to present the thermodynamic models that are prevailing in the study of multivalent interactions, which characterize protein–carbohydrate interactions. Mammen et al. showed the kinetics of multivalence interactions to be dependent on the thermodynamics of multivalent interactions and in particular minimizing entropic contributions (penalties) to the Gibbs free energy [2]. The influence of the linker molecule used to present the ligands or receptors in a multivalent fashion is shown to be crucial. Linker rigidity (and flexibility) and the mode of presenting their binding epitope are vitally important for controlling or maximizing enthalpy changes and minimizing entropic penalties to the Gibbs free energy, which in turn maximizes binding affinity at the individual and global binding events. The second model, proposed by Kramer and Karpen [38] and applied by Gargano et al. [39] and Mulder et al. [41], investigates the influence of the first binding event on following binding events. In this case, the kinetics of multivalent interactions are very much dependent on the statistics of the system and concentrations of interacting bodies. Due to this, there are no extrapolations or conclusions relating to the thermodynamics of the systems studied. Again, in this model, the nature of the linker molecule used is crucial for invoking multivalent interactions. In both models, several assumptions were made. Firstly, the multivalent receptors do not exhibit cooperativity and that the multiple interactions are independent, equal (of the same motif) and monotopic. It has been assumed that the multivalency originates from the architectural nature of the receptor being presented as several sub-units each with a ligand binding site.
Cooperativity occurs frequently in biological systems and often involves structural changes in the receptor itself in order to alter the binding of subsequent ligands. This must also be taken into account when designing a multivalent receptor so that the first interaction occurs in a manner which allows the second ligand to adopt the most appropriate position to interact with the new, “modified” receptor. So far we have discussed cases of architectural multivalence; multiple copies of receptor sub-units bound in an organized symmetric manner. In other cases of multivalence, the thermodynamic and effective concentration arguments presented above would change dramatically.
A wide range of multivalent architectures is available, with an equally wide variety of coupling methodologies. Therefore, the toolbox for studying multivalence and the cluster-glycoside effect is very diverse and constantly evolving. As well as providing control over valence; positioning, spacing and presentation of the saccharide moieties can also be controlled. The introduction of multiple functionalities for multiple targets is also possible. By taking advantage of particular properties of a scaffold, one can design molecular, macromolecular and supermolecular tools with a wide range of potential applications. Of course, the multivalent scaffold and functionalities used must reflect the properties of the multivalent receptor to be studied.
Among the different synthetic multivalent architectures, the tunability of ligand valency/presentation density and the potential for multiple functionalities offered by gold nanoparticles would allow them to provide valuable insights into the molecular basis which are underlined in the expression of the cluster glycoside effect. Taking into consideration the two arguments put forward for explaining this effect; thermodynamic enhancement and effective concentrations, complementarity between ligands presented on a multivalent scaffold corresponding to the distances between receptor binding sites is clearly an important factor for inducing multivalent interactions. Large inter-ligand distances would induce monovalent-like thermodynamics and concentration-dependent kinetics, favoring the occurrence of multiple intermolecular associations which in turn may lead to aggregation. Smaller inter-ligand distances would favor multivalent intramolecular interactions both thermodynamically and with reference to the effective concentration. At very small inter-ligand distances, however, the two models would disagree. Kinetics dependent on the effective concentration would predict a further increase in intramolecular association whereas thermodynamics would imply unfavorable steric interactions, reducing the occurrence of intramolecular interactions (but not necessarily increasing multiple intermolecular binding events either). Therefore, a combination of both models may be necessary to fully explain the glycoside cluster effect and its implications in experimental observations, particularly at very small inter-ligand distances.
Notes
Con A: Concanavalin A (Con A) is a leguminous lectin from the jack-bean Canavalia ensiformis. It adopts several isoforms depending on the environmental conditions. At a pH < 6, it exists as a homodimer whereas above pH 7 it exists as a homotetramer (or rather a dimer of dimers) with inter-binding site distances of ∼70 Å as seen in Fig. 2A [24]. Between pH 6–7, the lectin is in equilibrium between dimer and tetramer. The monomeric weight is 25.5 kDa, and each monomer contains one carbohydrate recognition domain and two metal binding sites. Con A is a C-type lectin [109] requiring the presence of Ca2+ ions for activity and thus one Ca2+ ion occupies one of the metal binding sites. The second metal binding site is reserved for a transition metal such as Mn, Co, Ni or Cd, all in the oxidation state II [110]. Due to the steric requirements imposed by several amino acids which form the topology of the binding pocket, Con A is specific for α-pyranose forms of mannosides and glucosides (which includes GlcNAc), which are C2 epimers of each other, with binding occurring between hydroxyl groups of the 3, 4 and 6 positions of the pyranose ring [111,112]. More specifically, cooperative hydrogen bonding occurs between the aforementioned hydroxyl groups and the binding pocket as well as hydrophobic stacking and Van der Waals interactions. Hydrogen bond networks are also formed which are mediated by water molecules. Indeed, five structural water molecules are displaced upon monosaccharide binding [111] (and references therein). Affinity for the mannoside is approximately six times greater than for the glucoside, (Ka of 1.2 × 104 M−1 and 2 × 103 M−1 for Me-α-d-mannose and Me-α-d-glucose, respectively) and 12 times greater than GlcNAc (1 × 103 M−1) [45,113].
PA-IL: one of several lectins from Pseudomonas aeruginosa, LecA (PA-IL), like BclA is a soluble C-type bacterial lectin implied in bacterial invasion, surface adhesion, biofilm formation and antibiotic resistance. Again, this lectin, in combination with other P. aeruginosa lectins, is known to play an important role in pulmonary infections of patients suffering from cystic fibrosis [27]. The lectin has a monomeric molecular weight of ∼12.8 kDa, which associate to form tetramers under physiological conditions, with an elongated square-planar architecture as shown in Fig. 2C [25,114]. The inter-binding site distances were measured as 29, 78 and 72 Å in the x, y and diagonal axes, respectively. Like BclA, PA-IL has a strict specificity, however, in this case for d-galactose and d-galactose-containing oligosaccharides. PA-IL is also known to bind GalNAc, although with a lower affinity, and adenosine [115,116]. Each monomer contains one carbohydrate binding domain and one Ca2+ ion. The crystal structure of PA-IL co-crystallized with galactose shows the formation of hydrogen bonds between hydroxyls in the 2, 3 and 4 positions whilst the hydroxyls of the 3 and 4 positions are also involved in complexing the Ca2+ ion. Hydrophobic interactions and bridging structural water molecules are also observed [25]. For d-galactose, PA-IL exhibits a Ka of 3.4 × 104 M−1 as calculated by equilibrium dialysis [117].
BclA: the lectin from Burkholderia cenocepacia, known as Burkholderia cenocepacia lectin A (BclA) is a recently-characterized soluble bacterial lectin implied in bacterial invasion and biofilm formation, particularly in patients suffering from cystic fibrosis [26]. This lectin is significantly smaller than Con A, forming homodimers in physiological conditions, with each monomer exhibiting a molecular mass of 13.8 kDa and an inter-binding site distance of ∼40 Å (Fig. 2B). Like Con A, BclA is a C-type lectin and each monomer contains one carbohydrate recognition domain which includes two Ca2+ ions directly involved in ligand binding. It is specific for mannose and is strictly limited to d-mannose and d-mannose-containing oligosaccharides. This specificity is related to a particular axial/equatorial arrangement of hydroxyls in the 2, 3 and 4 positions for direct coordination to the two Ca2+ ions and the topology of the carbohydrate binding site. The hydroxyls of positions 2, 3, 4 and 6 are also involved in hydrogen bonds to amino acids of the binding sites. Hydrophobic and aromatic interactions are also evident. For Me-α-d-Man, BclA exhibits a Ka ∼25 times stronger than that of Con A (Ka of ∼3 × 105 M−1).
Acknowledgements
The authors wish to thank the GlycoGold research training network, a part of the sixth research framework program of the European Union, contract number MRTN-CT-2004-005645, for financial support. Financial support from the CNRS and ESRF are also acknowledged.