

1 Introduction
Due to their unique position on the periodic table, actinoids such as uranium present several challenges to controlling reactivity through ligand design [1]. These challenges include the relatively large ionic radii that are encountered in the actinoid series, and the availability of both d- and f-orbitals to engage in metal-ligand interactions. In the case of uranium, the ionic radii span a wide range (1.03 Å (U3+) − 0.73 Å (U6+), for coordination number of 6), while coordination numbers range from 3 to at least 9 [2,3]. Controlling the coordination sphere of uranium across multiple oxidation states while minimizing structural reorganization may be effected by enforcing steric constraints about the metal center and by managing the degree of coordinative saturation provided by the ancillary ligand set(s) [4–7].
Amides are one widely explored ancillary ligand class that has been applied to uranium with great success [8]. Uranium amides have been useful for demonstrating novel stoichiometric reactions, and several applications to catalysis have been demonstrated [9–11]. We have pursued low-coordinate uranium complexes supported by N-alkylanilide ligands of the form N[R]Ar (R = t-Bu, C(CD3)2CH3, 1-adamantyl; Ar = 3,5-Me2C6H3). These bulky monodentate ligands have assisted in stabilizing an example of a uranium-silicon single bond [12] and the first monometallic uranium nitride complex [13], as well as examples of η6-arene coordination [14] and cooperative reduction of dinitrogen [15].
Herein, we present several uranium-containing complexes featuring a new bidentate anilide ligand. We show that this new ligand is capable of supporting both homoleptic and heteroleptic uranium(III) and uranium(IV) derivatives, and we provide crystallographic evidence suggesting that this new ligand has properties that can render a central uranium ion sterically well-protected.
2 Results and discussion
2.1 Synthesis of HN[R]ArMeL and its lithium and potassium derivatives
We sought to develop a bidentate anilide ligand that provides access to a six-coordinate uranium(III) derivative. We envisioned that such a system, given the proper conditions and steric loading, would allow for novel reaction chemistry incorporating the reducing nature of uranium(III) and a limited ability to significantly expand the coordination sphere about the uranium center. Appending a simple dialkylamino residue at the 2-position of the aryl group in an N-alkylanilide would provide a monoanionic bidentate ligand suitable for achieving this goal. Accordingly, we prepared the ligand salts (Et2O)Li(N[R]ArMeL) (R = C(CD3)2CH3); ArMeL = 2-NMe2-5-MeC6H3) and K(N[R]ArMeL) in good yields via modifications of literature procedures [16,17]. Briefly, zinc reduction of 2-nitro-N,N-dimethyl-p-toluidine provided the primary aniline H2NArMeL in 70 % yield. Condensation of H2NArMeL with acetone-d6 gave the hexadeuterated imine (CD3)2C = NArMeL in 86 % yield. Treatment of (CD3)2C = NArMeLwith MeLi in Et2O followed by an aqueous workup led to the isolation of the secondary aniline HN[R]ArMeL in 91 % yield. Finally, treatment of HN[R]ArMeL with n-BuLi provided (Et2O)Li(N[R]ArMeL) in 89 % yield, while treatment of HN[R]ArMeL with KH provided KN[R]ArMeL in 92 % yield, both salts being obtained as white powders following standard workup.
2.2 Uranium(III) complexes supported by the N[R]ArMeL ligand
We found that the number of N[R]ArMeL ligands readily appended to a uranium(III) center through simple salt elimination reactions was dependent upon the counter-cation employed in the ligand precursor. This substitutional selectivity allowed us to access both bis(anilide) and tris(anilide) systems (Fig. 1).
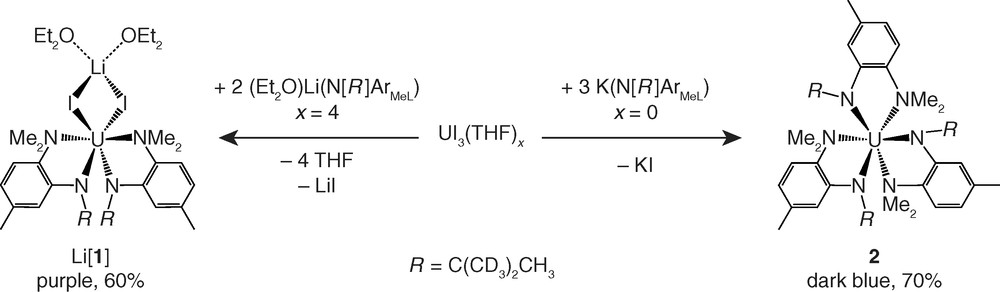
Synthesis of bis- and tris-anilide derivatives of uranium(III) incorporating the bidentate N[R]ArMeL ligand.
Treatment of UI3(THF)4 with (Et2O)Li(N[R]ArMeL) (2 equiv) in toluene resulted in formation of (Et2O)xLi[I2U(N[R]ArMeL)2] (Li[1]) as a dark purple powder in 60 % yield following separation from LiI and precipitation of the product from solution1. The 1H NMR spectrum of Li[1] in benzene-d6 consists of seven broad singlets found in a range spanning from +53 to −59 ppm, while the 2H NMR spectrum contains one broad feature at +53 ppm. In the absence of a vertical mirror plane, the two N-methyl groups of the N[R]ArMeL ligand are rendered inequivalent. Thus, the number of observed NMR features suggests that [1]1- maintains C2 or Cs symmetry in solution on the NMR timescale.
The reaction of solvate-free UI3 with K(N[R]ArMeL) (3 equiv) in thawing THF provided U(N[R]ArMeL)3 (2) in 70 % yield following separation from KI and crystallization from Et2O. The 1H NMR spectrum of 2 in benzene-d6 was found to be quite complex and indicative of more than one ligand environment present in solution, but its variable temperature 2H NMR spectrum was very informative (Fig. 2). At 20 °C, the 2H NMR spectrum of 2 in benzene-d6 consists of three broad peaks at +23.6, −9.0, and −25.0 ppm, with the latter two peaks integrating in an approximate 1:2 ratio. At 40 °C, all three features are significantly broadened, and by 60 °C they have coalesced into one broad resonance centered at +4.7 ppm. This behavior suggests that in solution at 20 °C, 2 exists in an equilibrium between two separate conformers: a C3-symmetric species that gives rise to a single peak at +23.6 ppm, and a Cs-symmetric species that accounts for the two features at −9.0, and −25.0 ppm. Raising the temperature of the system leads to more rapid interconversion of these two conformers relative to the NMR timescale, which is reflected by coalescence of the three NMR features at elevated temperatures.
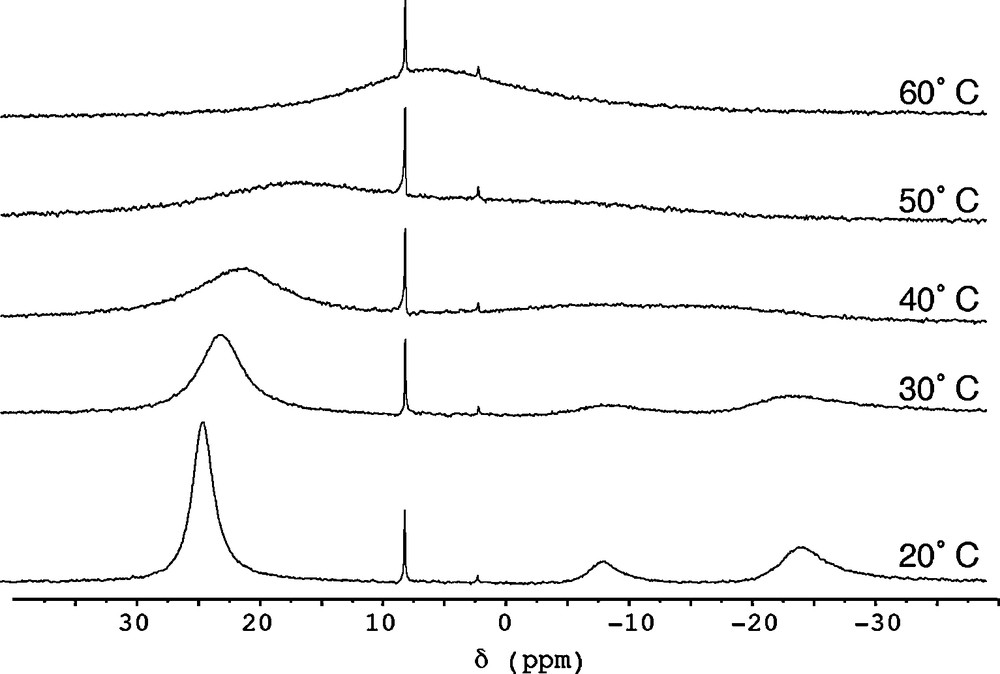
2H NMR spectra (76.8 MHz, benzene-d6) of 2 recorded from 20-60 °C.
2.3 Uranium(IV) complexes supported by the N[R]ArMeL ligand
Both Li[1] and 2 were found to be readily oxidized by 1e to their corresponding uranium(IV) derivatives (Fig. 3). Treatment of Li[1] with I2 (0.5 equiv) in Et2O led to precipitation of the neutral uranium(IV) complex I2U(N[R]ArMeL)2 as a bright red-orange powder that was isolated in 89 % yield by filtration. The 1H NMR spectrum of 1 in CDCl3 contains one set of seven well-resolved singlets ranging from +76.7 to −53.6 ppm, corresponding to a single N[R]ArMeL ligand environment, while the 2H NMR spectrum of 1 displays one peak at +76.7 ppm. Treatment of 2 with AgOTf (OTf = CF3SO3) in THF at −35 °C provided the uranium(IV) salt [U(N[R]ArMeL)3][OTf] ([2][OTf]) in 82 % yield following separation from Ag0 and subsequent standard workup. As with 1, the 1H NMR spectrum of [2][OTf] in CDCl3 consists of one set of seven singlets, indicative of a single ligand environment, however these spectroscopic features span a much smaller range, from +7.76 to −0.87 ppm; the 2H NMR spectrum of [2][OTf] in CHCl3 consists of a single broad resonance at −0.35 ppm. Not conveyed by the NMR spectra of [2][OTf] is the disposition of the triflate anion, whether it resides in the inner or outer coordination sphere of the uranium ion.
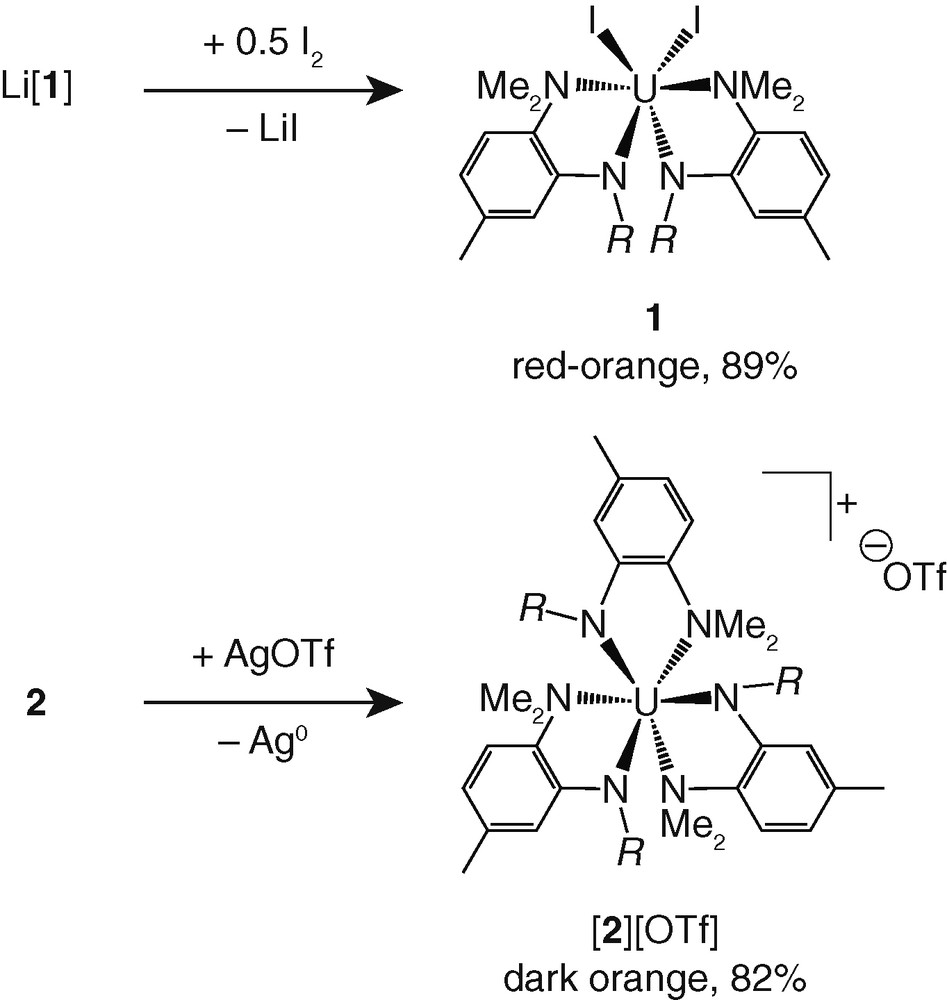
One-electron oxidation reactions with Li[1] and 2.
2.4 X-ray crystallographic investigations
To further understand the coordination mode adopted by the N[R]ArMeL ligand, we turned to single-crystal X-ray crystallography. Crystallization of Li[1] from an Et2O/THF mixture at −35 °C provided crystals of (Et2O)(THF)Li(μ-I)2U(N[R]ArMeL)2 ((Et2O)(THF)Li[1]), while crystals of 1, 2, and [2][OTf] were readily obtained by standard crystallization methods. The structural models obtained from our crystallographic investigations are presented in Fig. 4, and selected average interatomic distances are compiled in Table 1. In all four cases, the uranium centers are nominally six-coordinate, and the N[R]ArMeL ligands assume the expected bidentate coordination mode. The U-Nanilide distances fall in a range that is consistent with other reported uranium-anilide complexes [12–15,18], and the U-Namino distances are typical as well [19]. One noteworthy feature of all four structures is the interactions between the uranium center and the π-system of the anilide aryl backbone. One structural parameter that may be used to illustrate this interaction is the envelope angle (ϕenvelope), defined as the obtuse dihedral angle created by uranium center projecting out of the co-planar ligand-based N-C-C-N array [20]. The average values of ϕenvelope in (Et2O)(THF)Li[1] and 1 differ by less than one degree, while ϕenvelope in 2 to [2][OTf] differ by over 20°. This dramatic difference may be viewed as a consequence of charge separation in [2][OTf]. In all cases, the U-Cphenylene distances fall in a range common for the interaction between a uranium ion and a neutral arene fragment [21]. These interactions are frequently observed in uranium-anilide complexes, and are representative of fairly weak intramolecular interactions that nonetheless assist in stabilizing the central ion.
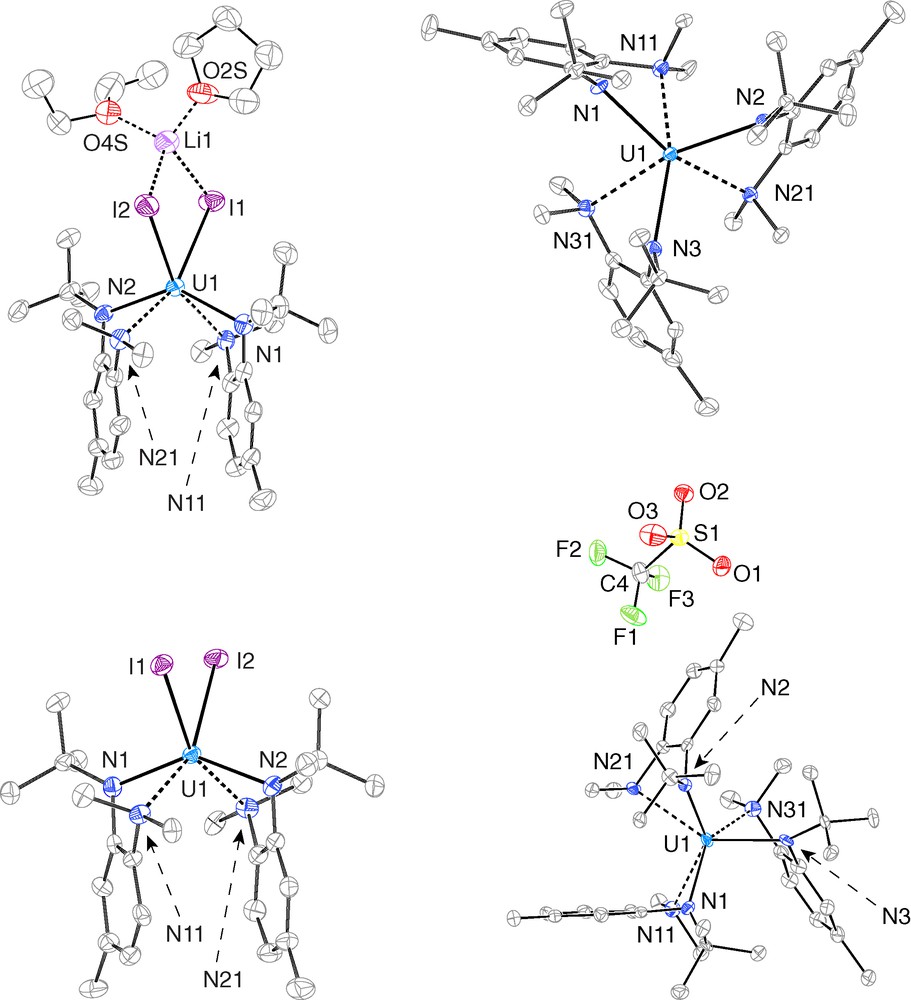
ORTEP renderings of (Et2O)(THF)Li[1] (top-left), 1 (bottom-left), 2 (top-right), and [2] [OTf] (bottom-right) generated by PLATON [26]. Ellipsoids are displayed at 50 % probability; hydrogen atoms and co-crystallized solvent molecules have been omitted for clarity.
Averaged key interatomic distances (Å) and envelope angles (°) in uranium complexes featuring the N[R]ArMeL ligand.
| Molecule | U–Nanilide | U–Namino | U–I | U–Cphenylene | ϕenvelope |
| (Et2O)(THF)Li[1] | 2.368 | 2.667 | 3.213 | 3.108 | 124.76 |
| 1 | 2.227 | 2.594 | 3.0976 | 2.970 | 123.13 |
| 2 | 2.467 | 2.752 | – | 3.292 | 135.21 |
| [2][OTf] | 2.242 | 2.723 | – | 2.919 | 115.02 |
Both (Et2O)(THF)Li[1] and 1 exhibit highly distorted octahedral coordination about the central uranium ion, with the iodide ligands occupying cis-equatorial sites. The uranium centers in (Et2O)(THF)Li[1] and 1 may alternatively be viewed as pseudo-tetrahedrally coordinated by N1, N2, I1, and I2, with the amino nitrogens N11 and N21 acting as additional capping ligands. The U-I distances in (Et2O)(THF)Li[1] and 1 are typical [19], and the ca. 0.13 Å decrease in the average U-I distance on going from (Et2O)(THF)Li[1] to 1 tracks well with difference in ionic radii between U3+ and U4+ (0.14 Å). Indeed, very little structural reorganization is observed upon proceeding from (Et2O)(THF)Li[1] to 1.
The primary coordination sphere in 2 is best described as a distorted trigonal anti-prism, with one trigonal plane comprising the anilide nitrogens and the other comprising the amino nitrogens. The primary coordination sphere in [2][OTf] is closer to trigonal prismatic, with the the trigonal planes defined as in 2. Of note is the fact that the triflate anion in [2][OTf] does not coordinate the uranium center. Visualizing a space-filling model of the [2]+ ion provides insight into why the triflate ion in [2][OTf] is relegated to the outer coordination sphere (Fig. 5). The uranium center in [2]+ is fully encapsulated in a hydrocarbyl sheath created by the three N[R]ArMeL ligands. In particular, the R and NMe2 residues interdigitate, and in doing so block off what might otherwise be accessible avenues for ligand coordination. Neutral 2 displays a similar degree of steric encumbrance at the uranium center, suggesting that it may prove challenging to utilize 2 for reaction chemistry based on inner-sphere redox activity.
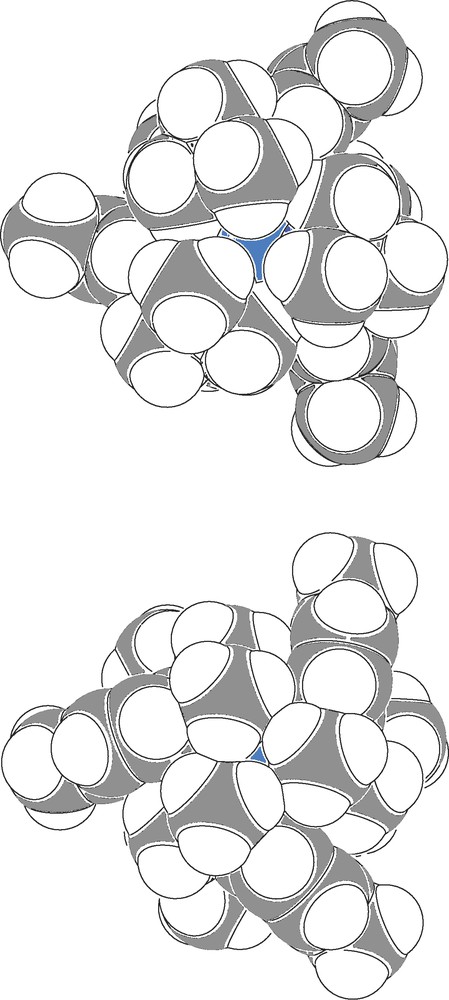
Two space-filling representations of the [2]+ ion (C, gray; H, white; N, blue; U, turquoise) generated by PLATON [26]: top, viewed looking down at the plane defined by N1, N2, and N3; bottom, viewed looking down at the plane defined by N11, N21, and N31.
3 Conclusion
Herein, we have described two new uranium amide systems based on the bidentate anilide ligand N[R]ArMeL. Both bis- and tris-anilide complexes of uranium(III) have been obtained, and the 1e oxidation of these compounds to the corresponding uranium(IV) complexes has been demonstrated. X-ray diffraction experiments confirm the bidentate nature of the N[R]ArMeL ligand, and in the case of the tris-anilide derivative reveals a central uranium ion that is subject to a high degree of steric congestion. The synthesis of this new chelating ligand is modular with respect to varying the alkyl and amino substituents, and our attention now turns to developing similar chelating ligands with steric properties amenable to accessing seven-coordination at uranium.
4 Experimental section
4.1 Methods and materials
All manipulations were carried out at room temperature (23 °C) under an atmosphere of purified dinitrogen in a Vacuum Atmospheres MO-40 M glove box or with a vacuum manifold using standard Schlenk techniques. Celite 545 (EM Science) and 4 Å molecular sieves were dried via storage in a 225 °C oven for 24 h followed by complete desiccation under dynamic vacuum at 210 °C for 48 h prior to use. Solvents were purified using a commercial Glass Contour solvent purification system constructed by SG Water USA (Nashua, NH USA), and were stored over activated 4 Å molecular sieves prior to use. Chloroform-d, benzene-d6, and acetone-d6 were obtained from Cambridge Isotope Laboratories, Inc. (Andover, MA USA); chloroform-d and benzene-d6 were vacuum distilled from CaH2, degassed with two freeze-pump-thaw cycles, and stored over activated 4 Å molecular sieves prior to use. UI3(THF)4 and UI3 were prepared according to published procedures [22,23]. 2-nitro-N,N-dimethyl-p-toluidine was prepared by nitration of N,N-dimethyl-p-toluidine with NaNO2 and Ce(SO4)2 according to published methods [24]. 2-dimethylamino-5-methylaniline (H2NArMeL) was prepared by reduction of 2-nitro-N,N-dimethyl-p-touidine using a modification of a literature procedure [16]; H2NArMeL was converted to HN[R]ArMeL via a modified literature procedure [17]. The latter two procedures are described below. All other reagents were obtained from commercial suppliers and used as received. All glassware was oven-dried at a temperature of 225 °C prior to use.
Solution 1H, 13C, and 2H NMR spectra were recorded on Varian Mercury-300 or Varian Inova-500 spectrometers at 20 °C. All 1H and 13C NMR spectra were referenced internally to residual solvent (C6D5H in C6D6, 7.16 ppm; CHCl3 in CDCl3, 7.27 ppm). All 2H NMR spectra were referenced internally to naturally abundant solvent deuterium peaks. GC-MS data were collected using an Agilent 6890N network GC system with an Agilent 5973 Network mass selective detector and an Rtx-1 column from Restek (Bellefonte, PA USA). Combustion analyses were performed by Columbia Analytical Services, Inc. (Tucson, AZ USA).
4.1.1 Synthesis of H2NArMeL
In a 1 L, three-necked flask fitted with a reflux condenser, a mixture of 2-nitro-N,N-dimethyl-p-touidine (53.18 g, 0.295 mol, 1 equiv), NaOH(aq) (36.0 mL of a 20 % w/w solution), and ethanol (220 mL) was prepared and magnetically stirred. The mixture was heated to reflux using a heating mantle. Once a gentle boil was achieved, the mantle was removed and zinc (90 g) was added in small portions (ca. 5 g at a time). The rate of zinc addition was fast enough to maintain the gentle boil. Half way through the addition (ca. 40 g Zn), the reaction mixture began to reflux with great vigor (it is imperative to have an ice bath ready for this eventuality). The reaction mixture was cooled in an ice bath for ca. 10 min during which time the reaction mixture maintained a steady reflux. Once the mixture began to evolve heat less vigorously, the addition of zinc was continued at ambient temperature, again in small portions. The addition at this point was done with great care, as the reaction mixture was prone to refluxing after each addition. Once all of the zinc was added, the heating mantle was returned and the reaction mixture was refluxed for 1.5 h. During the course of the reaction, the color of solution changed from bright red to dull brown. The hot reaction mixture was filtered through a bed of Celite on a sintered glass frit to remove excess zinc and any insoluble by-products. The filter cake was washed with ethanol (2 × 100 mL). The filtrate was taken to near dryness using a rotary evaporator. The dark brown residue was taken up in deionized water (500 mL) and extracted with diethyl ether (5 × 200 mL). The organic fractions were combined and dried with MgSO4. The mixture was then gravity filtered and the ether removed from the filtrate using a rotary evaporator. The dark brown oil that remained was distilled under reduced pressure (< 1 torr, 55 °C) to give the product as a nearly colorless oil (30.95 g, 0.206 mol, 70 %). 1H NMR (500 MHz, benzene-d6): δ = 6.84 (d, 2JHH = 10 Hz, 1H, 3- or 4-ArMeLH), 6.58 (d, 2JHH = Hz, 1H, 3- or 4-ArMeLH), 6.29 (s, 1H, 6-ArMeLH), 3.60 (br s, 2H, H2N)), 2.46 (s, 6H, N(CH3)2), 2.18 (s, 3H, ArMeLCH3) ppm. 13C{1H} NMR (75.5 MHz, benzene-d6): δ = 141.76 (s, ArMeL), 138.27 (s, ArMeL), 133.51 (s, ArMeL), 119.20 (s, ArMeL), 118.92 (s, ArMeL), 116.00 (s, ArMeL), 43.64 (s, N(CH3)2), 21.07 (s, ArMeLCH3) ppm. GC/MS: 150 (M·+) m/z.
4.1.2 Synthesis of (CD3)2C = NArMeL
A colorless solution of H2NArMeL (15.0 g, 0.100 mmol) and acetone-d6 (ca. 50 mL) was stored over 4 Å molecular sieves at 5 °C for 3 d. The solution was then transferred onto fresh 4 Å molecular sieves, and the used sieves were washed with acetone-d6. The washings were added to the bulk mixture, which was then stored at 5 °C for another 5 d. During this time, the solution obtained a faintly red color. The solution was decanted from the sieves, the sieves were then washed with acetone-d6, and the washings combined with the bulk mixture. Unreacted acetone-d6 was recovered by vacuum transfer, leaving behind spectroscopically pure (CD3)2C = NArMeL (16.93 g, 86.2 mmol, 86 %) as a red oil. If desired, the oil may be further purified by vacuum distillation at ca. 50 °C, but it is of sufficient purity to be taken on to the next step without any significant impact on yield. 1H NMR (500 MHz, benzene-d6): δ = 6.81 (mult, 2H, 3- and 4-ArMeLH), 6.53 (s, 1H, 6-ArMeLH), 2.57 (s, 6H, N(CH3)2), 2.20 (s, 3H,ArMeLCH3), 1.94 (mult, 0.15H, residual N = C(CH3)2), 1.44 (mult, 0.15H, residual N = C(CH3)2) ppm. 13C{1H} NMR (126 MHz, benzene-d6): δ = 167.38 (s, N = C(CD3)2), 145.93 (s, ArMeL), 141.73 (s, ArMeL), 131.66 (s, ArMeL), 124.56 (s, ArMeL), 121.39 (s, ArMeL), 118.25 (s, ArMeL), 42.85 (s, N(CH3)2), 27.86 (mult, N = C(CD3)2), 21.15 (s, ArMeLCH3). 2H NMR (76.8 MHz, benzene): δ = 1.94 (s, N = C(CD3)2), 1.44 (s, N = C(CD3)2) ppm. GC/MS: 160 (M − 2CD3) m/z.
4.1.3 Synthesis of HN[R]ArMeL
A solution of (CD3)2C = NArMeL (26.56 g, 0.135 mol, 1 equiv) in Et2O (50 mL) was added slowly to a stirred thawing solution of MeLi (200 mL, 1.6 M in Et2O, 0.320 mol, 1.6 equiv). The mixture assumed a yellow color and became cloudy. After stirring the mixture for ca. 14 h, the mixture was quenched by slow addition to an ice/water slurry (400 mL). The organic layer was separated and the aqueous layer was extracted with Et2O (3 × 200 mL). The combined organic fractions were dried over Na2SO4. Volatile materials were removed with the aid of a rotary evaporator, leaving a pale green oil that was then subjected to simple distillation under dynamic vacuum. The product was obtained as a yellow oil that distills at 60 °C under dynamic vacuum (25.97 g, 0.122 mol, 91 %). 1H NMR (300 MHz, benzene-d6): δ = 6.93 (d, 2JHH = 6 Hz, 1H, 3- or 4-ArMeLH), 6.84 (s, 1H, 8-ArMeLH), 6.56 (d, 2JHH = 8 Hz, 1H, 3- or 4-ArMeLH), 5.11 (br s, 1H, NH), 2.45 (s, 6H, N(CH3)2), 2.20 (s, 3H, ArMeLCH3), 1.30 (s, 3H, C(CD3)2CH3 ppm. 13C{1H} NMR (125.8 MHz, benzene-d6): δ = 142.85 (s, ArMeL), 139.48 (s, ArMeL), 134.40 (s, ArMeL), 120.26 (s, ArMeL), 117.41 (s, ArMeL), 114.43 (s, ArMeL), 50.01 (s, C(CD3)2CH3), 44.87 (s, N(CH3)2), 30.31 (s, C(CD3)2CH3), 23.30 (s, ArMeLCH3) ppm. GC/MS: 212 (M·+) m/z.
4.1.4 Synthesis of (Et2O)Li(N[R]ArMeL)
n-BuLi (48.0 mL, 1.6 M in hexane, 76.83 mmol, 1.3 equiv) was slowly added to a stirred solution of HN[R]ArMeL (12.55 g, 59.10 mmol, 1 equiv) in thawing n-pentane (80 mL). The mixture assumed a faintly yellow color and a small amount of white precipitate formed. The mixture was stirred for 30 min before Et2O (20 mL) was added. The mixture became homogeneous and was concentrated under reduced pressure to ca. 40 mL and was cooled in the glove box cold well, leading to the formation of a large amount of white precipitate. The product precipitate was isolated by filtering the cold mixture through a medium frit, then washing the retained solids with a small amount of cold n-pentane, and finally drying the solids under reduced pressure (15.38 g, 52.6 mmol, 89 %). 1H NMR (500 MHz, benzene-d6): δ = 6.79 (d, 2JHH = 8 Hz, 1H, 3- or 4-ArMeLH), 6.78 (s, 1H, 6-ArMeLH), 6.24 (d, 2JHH = 8 Hz, 1H, 3- or 4-ArMeLH), 3.04 (q, 4H, O(CH2CH3)2), 2.35 (s, 3H, ArMeLCH3), 2.20 (s, 6H, N(CH3)2), 1.56 (s, 3H, C(CD3)2CH3, 0.92 (t, 6H, O(CH2CH3)2) ppm. 13C{1H} NMR (75.5 MHz, pyridine-d5): δ = 154.59 (s, ArMeL), 139.63 (s, ArMeL), 133.00 (s, ArMeL), 117.10 (s, ArMeL), 113.15 (s, ArMeL), 101.53 (s, ArMeL), 52.01 (s, C(CD3)2CH3), 43.88 (s, N(CH3)2), 31.80 (s, C(CD3)2CH3), 23.50 (s, ArMeLCH3) ppm. 2H NMR (76.8 MHz, benzene): δ = 1.56 (s, C(CD3)2CH3) ppm.
4.1.5 Synthesis of K(N[R]ArMeL)
Solid KH (1.62 g, 40.4 mmol, 1.1 equiv) was added to a 200 mL recovery flask containing a stirred solution of HN[R]ArMeL (7.77 g, 36.6 mmol, 1 equiv) in THF (80 mL). The flask was capped with a septum and syringe needle was inserted into the septum to facilitate the escape of H2(g). The mixture was allowed to stir for 36 h, whereupon it was filtered through a Celite-padded frit to remove excess KH. Volatile material was removed under reduced pressure from the yellow filtrate, yielding a pale yellow solid. Toluene (60 mL) was added and subsequently removed under reduced pressure. The solid was then slurried in Et2O (30 mL) and n-hexane (80 mL). The total volume was reduced to ca. 30 mL by concentrating the mixture under reduced pressure, and the pale yellow precipitate was isolated by filtering the mixture through a medium frit. The solids were washed with n-pentane (2 × 20 mL) and dried under reduced pressure (8.40 g, 33.5 mmol, 92 %). 1H NMR (300 MHz, pyridine-d5): δ = 6.79 (d, 7 Hz, 1H, 3- or 4-ArMeLH), 6.18 (s, 1H, 6-ArMeLH), 5.97 (d, 8 Hz, 1H, 3- or 4-ArMeLH, 2.85 (s, 6H, N(CH3)2), 2.46 (s, 3H, ArMeLCH3), 1.65 (s, 3H, C(CD3)2CH3) ppm. 13C{1H} NMR (75.5 MHz, pyridine-d5): δ = 154.59 (s, ArMeL), 139.63 (s, ArMeL), 133.00 (s, ArMeL), 117.10 (s, ArMeL), 113.15 (s, ArMeL), 101.53 (s, ArMeL), 52.01 (s, C(CD3)2CH3), 43.88 (s, N(CH3)2), 31.80 (s, C(CD3)2CH3), 23.50 (s, ArMeLCH3) ppm. 2H NMR (76.8 MHz, pyridine): δ = 1.65 (s, C(CD3)2CH3) ppm.
4.1.6 Synthesis of Li[I2U(N[R]ArMeL)2]
Solid (Et2O)Li(N[R]ArMeL) (1.88 g, 6.44 mmol, 2 equiv) was added to a stirred suspension of UI3(THF)4 (2.922 g, 3.22 mmol, 1 equiv) in thawing toluene. The mixture was allowed to stir and warm to ambient temperature for 12 h, during which time the mixture was maintained under dynamic vacuum to remove solvent and other volatile materials. A purple residue remained, to which n-hexane (200 mL) was added. The mixture was filtered through a Celite-padded frit, and volatile materials were removed from the filtrate under reduced pressure. Et2O (20 mL) and n-hexane (50 mL) were added to the residue thus obtained, creating a solution. Volatile materials were removed under reduced pressure from this solution, and again Et2O (20 mL) and n-hexane (50 mL) were added and removed under reduced pressure. n-Pentane (20 mL) was then added, creating a purple suspension. The inner walls of the flask were scraped to dislodge adhered material, and the suspened solids were isolated by filtering the mixture through a medium frit. The solids were washed with n-pentane (10 mL) and then dried under reduced pressure, providing the product as a purple powder (1.77 g, 1.93 mmol, 60 %). Crystals of this material were grown from a solution Li[1] in a mixture of THF/Et2O stored at −35 °C. 1H NMR (300 MHz, benzene-d6), as [(Et2O)xLi][I2U(N[R]ArMeL)2]: δ = 53.00 (br s, 18H, C(CD3)2CH3), 31.60 (br s, 9H, N(CH3)(CH3), 16.17 (br s, 3H, ArMeLH), −3.94 (br s, 9H, ArMeLCH3), −9.09 (br s, 3H, ArMeLH), −27.45 (br s, 3H, ArMeLH), −59.52 (br s, 9H, ArMeLCH) ppm. 2H NMR (76.8 MHz, benzene): δ = 53.0 (br s, C(CD3)2CH3) ppm.
4.1.7 Synthesis of U(N[R]ArMeL)3
Solid K(N[R]ArMeL) (1.521 g, 6.072 mmol, 3.4 equiv) was added to a stirred suspension of UI3 (1.100 g, 1.777 mmol, 1 equiv) in thawing THF (80 mL). The resulting mixture was stirred for 2.5 h, over which time the mixture darkened from a purple-blue to black. The mixture was filtered through a Celite-padded frit to remove precipitated KI. Volatile materials were removed under reduced pressure from the filtrate, leaving behind a dark blue-black residue. Et2O (50 mL) and n-pentane (50 mL) were added to the residue, and the resulting mixture was filtered through a Celite-padded frit. Volatile materials were removed under reduced pressure from the filtrate, leaving behind a dark blue-black powder. The powder dissolved in Et2O (20 mL) and the resulting solution was filtered through a plug of Celite. The filtrate was stored at −35 °C, resulting in the product depositing on the walls of the storage flask as dark blue-black microcrystals. The product was isolated in three crops by removing the mother liquor by pipet and drying the microcrystals under dynamic vacuum (1.067 g, 1.223 mmol, 69 %). Single crystals of this material were grown from a solution of 2 in a mixture of THF/Et2O/n-pentane stored at −35 °C. 2H NMR (76.8 MHz, benzene-d6): δ = 23.6 (br s, C3-symmetric conformer), −9.0 (br s, Cs-symmetric conformer) and −25.0 (br s, Cs-symmetric conformer) ppm.
4.1.8 Synthesis of I2U(N[R]ArMeL)2
A thawing solution of I2 (0.081 g, 0.319 mmol, 0.5 equiv) in Et2O (5 mL) was added dropwise to a stirred thawing solution of Li[I2U(N[R]ArMeL)2] (0.587 g, 0.624 mmol, 1 equiv) in Et2O (6 mL), resulting in the immediate formation of a bright orange precipitate. The mixture was allowed to stir for 30 min before it was concentrated to half of its initial volume under reduced pressure. The precipitate was isolated by filtering the mixture through a medium frit. The solids were washed with n-pentane (10 mL) and dried under reduced pressure, yielding the product as a bright red-orange powder (0.507 g, 0.554 mmol, 89 %). Crystals of this material were grown from a solution of 1 in a mixture of CH2Cl2/THF/Et2O stored at −35 °C. 1H NMR (300 MHz, CDCl3): δ = 76.67 (s, 9H, C(CD3)2CH3), 30.21 (s, 3H, ArMeLH), 5.90 (s, 9H, ArMeLCH3), −10.60 (s, 9H, N(CH3)(CH3), −19.46 (s, 3H, ArMeLH), −36.21 (s, 3H, ArMeLH), −53.55 (s, 9H, N(CH3)(CH3)) ppm. 2H NMR (76.8 MHz, CHCl3): δ = 76.7 (s, C(CD3)2CH3) ppm.
4.1.9 Synthesis of [U(N[R]ArMeL)3][OTf]
A solution of AgOTf (0.0752 g, 0.280 mmol, 1 equiv) in cold THF (6 mL, −35 °C) was added to a stirred solution of U(N[R]ArMeL)3 (0.255 mmol, 0.280 mmol, 1 equiv) in cold THF (6 mL, −35 °C). The color of the mixture immediately went from purple-black to orange-brown. The mixture was allowed to stir for 1.5 h before it was filtered through a Celite-padded frit. The filter cake was washed with THF (20 mL) and volatile materials were removed from the combined filtrate under reduced pressure. Addition of an Et2O/n-pentane mixture created a suspension of a brown solid in a faintly brown supernatant solution. The suspended material was isolated by filtering the mixture through a medium frit and washing the isolated solids with n-pentane (5 mL) before drying the solids under reduced pressure. The product was thus obtained as dark orange precipitate (0.244 g, 82 %). Crystals of this material were grown from a THF solution of [U(N[R]ArMeL)3][OTf] layered with Et2O and stored at −35 °C. 1H NMR (500 MHz, CDCl3): δ = 7.76 (s, 3H, ArMeLH), 7.02 (s, 3H, ArMeLH), 6.81 (s, 3H, ArMeLH), 3.50 (s, 9H, N(CH3)(CH3), 3.26 (s, 9H, ArMeLCH3), −0.35 (s, 18H, NC(CD3)2CH3), −0.87 (s, 9H, N(CH3)(CH3) ppm. 2H NMR (76.8 MHz, CHCl3): δ = −0.35 (br s, C(CD3)2CH3) ppm.
4.2 X-ray crystallographic details
Low-temperature diffraction data were collected on a Siemens Platform three-circle diffractometer coupled to a Bruker-AXS Smart Apex charge-coupled device (CCD), performing φ- and ω-scans. The structures were solved by either direct methods or Patterson methods, in conjunction with standard Fourier difference techniques, and refined on F2 by full-matrix least-squares procedures. A semi-empirical absorption correction was applied to the diffraction data for all structures. All non-hydrogen atoms were refined anisotropically; all hydrogen atoms were placed at calculated positions and refined isotropically using a riding model. All software used for diffraction data processing and crystal structure solution and refinement are contained in the APEX2 v2008-3.0 program suite (Bruker AXS). The structure of (THF)(Et2O)Li[1]-0.5(Et2O) exhibits a disorder that switches the positions of the THF and Et2O molecules that coordinate the lithium cation; this disorder refined to 52 %. The refined structural model of 1 contained highly disordered solvent molecules in solvent-accessible channels for which no acceptable model was constructed. Consequently, the SQUEEZE routine [25] as implemented in PLATON [26] was used to remove the unassigned electron density. The refined model for [2][OTf]·2(THF) has the uranium-containing fragment placed in a PART -1 function. One consequence of this model is short non-bonded interatomic distances between the outer-sphere triflate and anilide residues in the unit cell adjacent to the triflate ion. It is presumed that the orientation of the uranium-containing fragment strictly alternates from one cell to the next, so no short interatomic contacts actually occur. Summaries of crystallographic data for complexes (Et2O)(THF)Li[1], 1, 2, and [2][OTf] are given in Table 2. Complete crystallographic details for these complexes are available in the form of Crystallographic Information Files (CIF) and can be obtained free of charge from the Cambridge Crystallographic Data Centre (depositions 761469-761472) via the Internet at http://www.ccdc.cam.ac.uk.datarequest/cif (or directly: Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336 033).
Crystallographic data for the structures presented in this work.
| (Et2O)(THF)Li[l]·0.5(Et2O) | 1 | 2 | [2][OTf]-2(THF) | |
| Empirical formulaa | C36H65I2LiN4O2.5U | C29H42I2N4U | C39H63N6U | C48H79F3N6O5SU |
| Formula weight (g/mol) | 1092.69 | 902.47 | 853.98 | 1147.26 |
| Temperature (K) | 100(2) | 100(2) | 100(2) | 100(2) |
| Wavelength (Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | Monoclinic | Monoclinic | Orthorhombic | Monoclinic |
| Space group | C2/c | C2/c | Fdd2 | P21/m |
| Unit cell dimensions (Å,°) | a = 34.656(4), α = 90 | a = 41.121(4), α = 90 | a = 24.8112(19), α = 90 | a = 11.1943(15), α = 90 |
| b = 16.3018(18), β = 105.405(2) | b = 9.7154(8), 96.114(2) | b = 64.566(5), β = 90 | b = 14.3832, β = 92.661(2) | |
| c = 15.9409(17), γ = 90 | c = 17.9839, γ = 90 | c = 9.5274(7), γ = 90 | c = 15.597(2), γ = 90 | |
| Volume (Å3) | 8682.3(16) | 7143.9(10) | 15,262(2) | 2508.6(6) |
| Z | 8 | 8 | 16 | 2 |
| Density (calculated) (Mg/m3) | 1.672 | 1.678 | 1.487 | 1.519 |
| Absorption coefficient (mm−1) | 5.195 | 6.289 | 4.288 | 3.338 |
| F(000) | 4240 | 3392 | 6896 | 1168 |
| Crystal size (mm3) | 0.15 × 0.13 × 0.05 | 0.16 × 0.11 × 0.05 | 0.25 × 0.05 × 0.05 | 0.27 × 0.08 × 0.05 |
| Theta range for collection (°) | 1.81 to 28.70 | 1.99 to 28.28 | 1.26 to 29.57 | 1.31 to 28.70 |
| Index ranges | −46 ≤ h ≤ 46 | −54 ≤ h ≤ 54 | −34 ≤ h ≤ 34 | −15 ≤ h ≤ 15 |
| −22 ≤ k ≤ 22 | −12 ≤ k ≤ l2 | −89 ≤ k ≤ 89 | −19 ≤ k ≤ 19 | |
| −21 ≤ l ≤ 21 | −23 ≤ l ≤ 23 | −13 ≤ l ≤ 13 | −21 ≤ l ≤ 21 | |
| Reflections collected | 88,114 | 62,723 | 76,743 | 48,855 |
| Independent reflections | 11,226 [R(int) = 0.0831] | 8853 [R(int) = 0.0770] | 10,716 [R(int) = 0.0837] | 10,716 [R(int) = 0.0745] |
| Completeness to θmax (%) | 100.0 | 100.0 | 100.0 | 100.0 |
| Absorption correction | Semi-empirical from equivalents | Semi-empirical from equivalents | Semi-empirical from equivalents | Semi-empirical from equivalents |
| Max. and min. transmission | 0.7812 and 0.5096 | 0.7439 and 0.4327 | 0.8142 and 0.4136 | 0.8509 and 0.4660 |
| Refinement method | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 11,226/672/527 | 8853/0/310 | 10,716/1/434 | 6727/179/595 |
| Goodness-of-fitb | 1.006 | 1.072 | 1.022 | 1.023 |
| Final R indices [I > 2σ(I)]c | ||||
| R indices (all data)c | ||||
| Largest diff. peak and hole (e Å−3) | 0.980 and −1.491 | 1.317 and −1.748 | 1.307 and −0.814 | 2.037 and −1.065 |
a All 2H atoms refined as 1H.
b .
c
Acknowledgements
We thank the U.S. National Science Foundation (CHE-0724158) for supporting this work. Financial support for EMT was provided by the Amgen Scholars program (http://www.amgenscholars.eu) with direction and assistance provided by the Undergraduate Research Opportunities Program at MIT.
1 The protocol for the synthesis and isolation of (Et2O)xLi [1] results in a variable and non-stoichiometric amount of Et2O binding to the lithium ion, a consequence of drying the isolated material under dynamic vacuum. The amount of Et2O that remains bound to the lithium ion after drying the isolated material has not been rigorously quantified, but it is assumed to be a small fraction of the total molecular weight and it is disregarded in subsequent calculations using Li[1].