

1 Introduction
The continued interest in carbohydrates is due to the chemical and the biological interest of these molecules. Their structural diversity, together with heterocyclic ring sizes and the variations in their sequences, position and configuration of the linkages among monosaccharide units can lead to libraries containing large numbers of carbohydrate-based structures [1]. From the biological point of view, carbohydrates play a very important role in cell–cell recognition processes: indeed, many pathogens use carbohydrate-binding proteins to attach themselves to cell surfaces and initiate diseases. Oligosaccharides and sugar molecules may also have potential therapeutic values against many of these diseases. However, advances in this field suffer from the fact that, unlike peptides and oligonucleotides, solid-phase synthesis of oligosaccharide-based libraries has not yet achieved great enough efficiency [2].
Bearing in mind the growing importance of glycobiology, the biological functions of carbohydrates and the problems associated in the synthesis of carbohydrate-based libraries, intensive attention is being devoted to the synthesis and biological applications of carbohydrate mimetics that may find useful applications in new drug discoveries and in the development of new materials. Saccharides have been shown to be suitable scaffolds for this purpose, on account of their high density of functional groups and their availability as chiral building blocks.
In the search for carbohydrate mimetics, particular attention has been devoted to the design and development of hybrid molecules, including sugar amino acids (SAA), a class of hybrid carbohydrate derivatives bearing amino and carboxylic acid functionalities, which can be classified according to the position of the amino acid moiety on the cyclic polyol [3]. Among them, SAAs arising from the direct incorporation of the amino acid moiety on the cyclic carbohydrate scaffold have emerged as a new class of promising hybrids that have played an important role in drug design and development. Their rigid furan and pyran rings make them ideal non-peptidic scaffolds for incorporation into peptides or peptidomimetics in order to induce conformational restrictions and enhance metabolic stability in pharmacologically active peptides [3]. In addition, the presence of several stereogenic centers on these rings can be exploited for the creation of chemical diversity and protection or unprotection of hydroxy substituents’ open opportunities for access to hydrophobic or hydrophilic peptidomimetics [4].
Carbasugars are another family of synthetic and naturally occurring carbohydrate mimics that are attracting great attention among chemists and biochemists (for a recent review on carbasugars, see [5]). They are polyhydroxylated five- or six-membered carbocyclic structures topologically similar to natural sugars, particularly in the arrangement of their hydroxy groups, but they have the ring oxygen replaced by a methylene group. Because of their lack of an easily hydrolyzable glycosidic function, these sugar derivatives are much more stable towards hydrolysis.
In connection with the present interest in carbasugars, five and six membered carbasugar amino acids, particularly those such as 2-α or 2-β (Fig. 1), which have the carboxyl and the amino groups directly linked to rings, are receiving growing attention. Given that they have the similar relationship to SAAs as between sugars and carbasugars, they may be regarded as suitable mimetics of SAA where the lack of an anomeric position allows avoiding the anomerization processes present in sugars and their hydrolysis by glycosidases (Fig. 1).
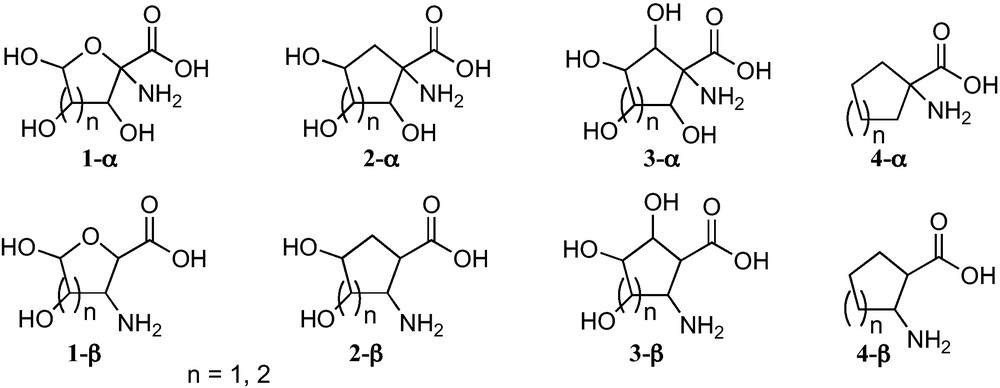
This microreview provides a perspective of work that has been carried out in our research group and by other research groups and comprises an overview of the synthesis of five and six membered sugar and carbasugar α- and β-amino acids from carbohydrates (Fig. 1) [6]. Although some previous reviews covering different aspects of sugar [3b,4,7] or carbasugar [8] amino acids have appeared, including a recent compendium of SAAs [9], this is the first common presentation in a review of the synthetic strategies developed for the preparation of these complementary scaffolds from carbohydrates. The main advantage of SAAs is their ready preparation from sugars, but they do, however, sometimes suffer the inconvenience of anomeric equilibria and their easy hydrolysis. These problems are not present in carbasugar amino acids, which show two relevant additional advantages:
- • as their structure includes a carbocyclic ring instead of a furanose or a pyranose ring, they keep a closer topological relationship with the corresponding unsubstituted cycloalkyl α- an β-amino acids 4-α and 4-β, respectively, which are of great present interest in peptidomimetics [10];
- • they open more opportunities for the creation of chemical diversity. The chemical diversity provided by carbasugar amino acids 2-α or 2-β can be easily increased by the consideration of 3-α or 3-β carbasugar amino acids.
Oxetane-3-amino-2-carboxylic acids are not covered, because at present they have received much less attention [11]. In fact, an only natural member of this family of oxetane β-amino acids, the antibiotic oxetin (5), has been reported. Regarding synthetic studies, several sugar-derived C,N-protected analogues 6a, 6b and 7 were prepared and the corresponding hexamers showed a remarkable propensity to fold in novel 10-helical conformations [12] (Fig. 2).
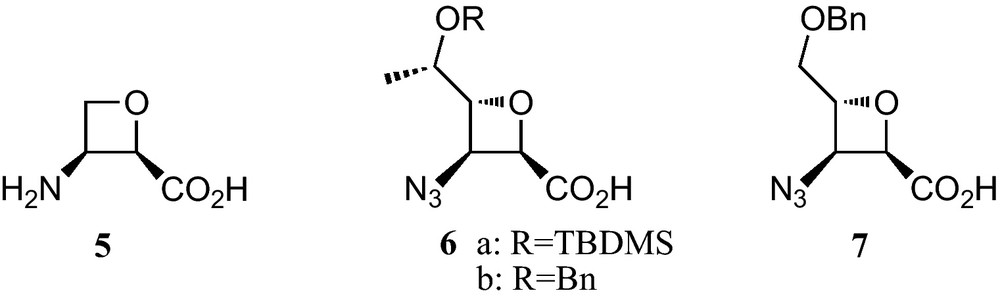
Sugar and carbasugar γ-amino acids are neither covered in this review, because at present, carbasugar γ-amino acids have not been prepared from carbohydrates. Sugar and carbasugar δ-amino acids are neither considered, because carbasugar δ-amino acids have not yet been reported.
2 Furanoid and pyranoid sugar α-amino acids
SAAs [7b,7c] occur naturally as subunits of oligosaccharides (neuraminic acid), in cell walls of bacteria (muraminic acid), and in some antibiotics [13]. In addition, since the first reported synthesis of a SAA [14], a vast number of SAAs have been prepared for their use as glycosidase inhibitors, starting materials in polymer and natural product synthesis, and as a new class of building blocks for peptide scaffolds and conformational restrained peptidomimetics [15].
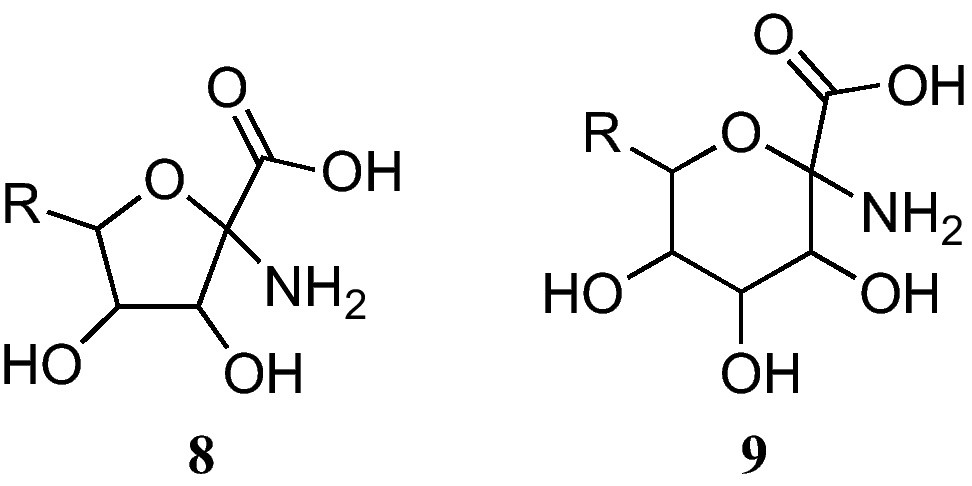
The furanoid sugar α-amino acids (8) at present reported bear a fused glycine at the anomeric position (Fig. 3) [9]. On the other hand, pyranoid sugar α-amino acids 9 have also been described [9].
Initial interest in furanoid sugar α-amino acids was closely connected with the large attention that spironucleosides attracted with the discovery in 1990 of (+)-hydantocidin (10, Fig. 4), the first natural spiroribofuranose isolated from the culture broth of Streptomyces hydroscopicus [16]. It has a unique structural feature consisting of a spirohydantoin at the anomeric position of a ribofuranose and displays both a plant growth regulatory and strong herbicidal activities against weeds with no toxicity to microorganisms and mammals. 1-Epihydantocidin as well as all the other stereoisomers and some derivatives and carbocyclic analogues of (+)-hydantocidin were also prepared in order to study structure–activity relationships [17]. Spirohydantoins of sugars other than ribose were also synthesized in the hope of discovering novel interesting biological properties [18].
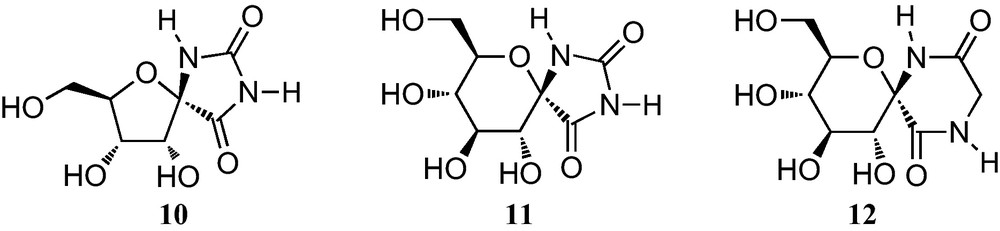
The discovery, in 1995, that the glucopyranose analogue 11 is a powerful inhibitor of glycogen phosphorylase, has stimulated the synthesis of hexopyranose analogues of spiro-hydantoins, as well as some hexofuranose analogues [19]. In addition, a wide range of anomeric diketopiperazines have also been prepared, including the spirodiketopiperazine of glucopyranose 12, which is a specific inhibitor of glycogen phosphorylase [20].
The interesting biological profile shown by these compounds prompted several groups to develop syntheses of these spironucleosides based on the previous preparation of the corresponding furanoid or pyranoid sugar α-amino acids. Therefore, only some representative examples of the synthetic strategies developed for this purpose are mentioned herein.
2.1 Synthetic approaches towards furanoid α-amino acids
Early work in this field by Fleet et al. consisted of the preparation of tetrahydrofurane α-SAA from 2-amino sugar lactones by an oxidative ring contraction (Scheme 1), which involves a one-pot oxidation of the amine 13 to its imine derivative 14 and subsequent opening of the lactone ring, followed by an intramolecular attack by the free OH group on the imine. This sequence resulted in the formation of a 4.8:1 anomeric mixture of compounds 15 and 16 bearing a furane ring and the α-amino acid moiety ([21] and references therein).
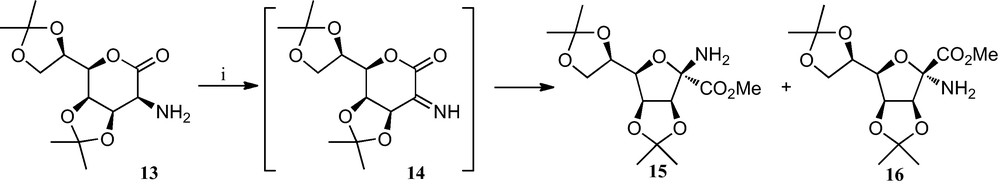
(i) Br2, Et3N, NaOAc, MeOH.
A slight modification of this strategy was later implemented for the preparation of furanoid sugar α-amino acid 20, the only component of this family of amino acids without a substituent at position C-4 (Scheme 2) [22]. Although it can be envisaged as being related to pentofuranoses, it was prepared by a Henry reaction of d-glyceraldehyde and ethyl nitroacetate, that allowed the synthesis of a 6:5 diastereomeric mixture of 3,4,5-trihydroxy-2-nitropentanoic acid esters, which were readily converted into the key furonolactones 17. Oxidation of this compound was followed by a spontaneous intramolecular attack by the free OH group on the resulting iminolactone 18, a process that provided a stereocontrolled route to the bicyclic lactone 19, which finally was efficiently opened to the parent amino acid 20.
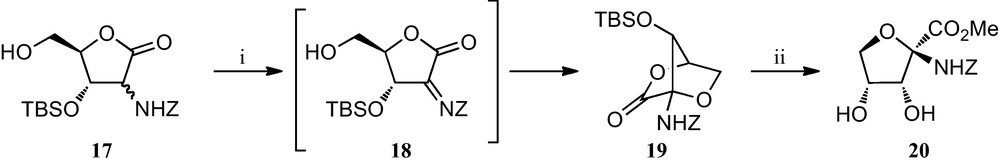
(i) NBS, CH3CN; (ii) (a) HCl, (b) TBAF.
A representative example of an alternative approach for the generation of the amino acid moiety of furanoid sugar α-amino acids, also developed by Fleet et al., is displayed in Scheme 3 [19d,23]. The rhamnose-derived sugar lactone 21a was transformed into the corresponding triflate 21b, which was directly subjected to a base-mediated ring contraction to obtain the furanosyl α-carboxylate 22. Radical bromination, followed by nucleophilic displacement of the bromine by an azido group, afforded the corresponding α-azido ester 23, which upon catalytic hydrogenation gave the α-amino ester 24, precursor of the corresponding α-amino acid.
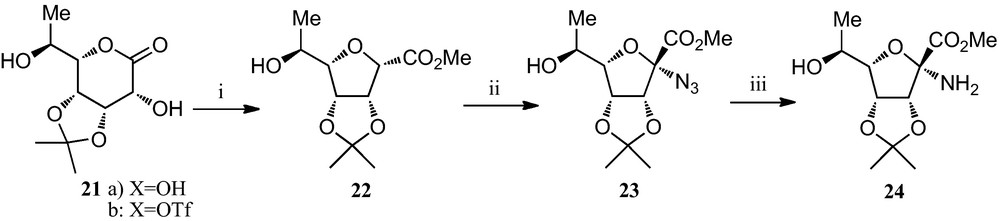
(i) (a) Tf2O, pyridine, CH2Cl2; (b) K2CO3, MeOH; (ii) (a) N-bromosuccinimide, (PhCO2)2, CC14; (b) NaN3, DMF; (iii) H2, Pd-black, EtOH.
A related approach (Scheme 4) consists of a two-step route to α-chloro-ulosonic esters 29 from lactones, involving the reaction of dichloroolefins with m-chloroperbenzoic acid in dichloromethane, followed by the substitution of chlorine in 29 by the azide ion, yielding the corresponding anomeric azido-esters 30, which may be useful intermediates for the preparation of anomeric amino acids and related compounds [24].
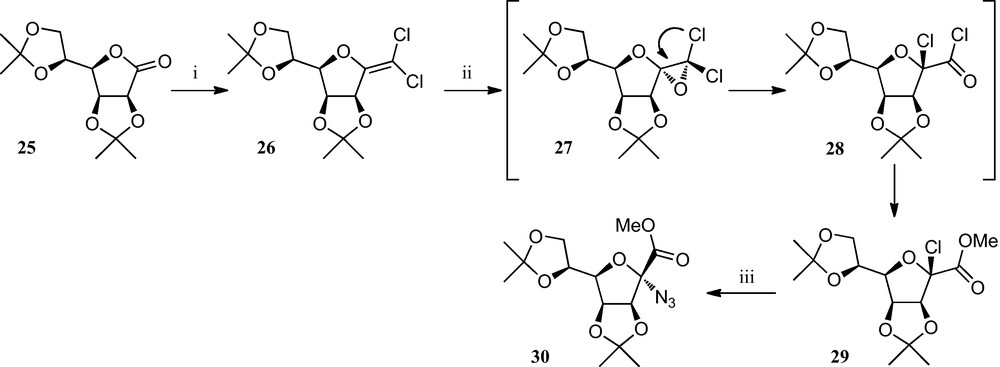
(i) [24a]; (ii) MCPBA, MeOH, hydroquinone, CH2Cl2; (iii) NaN3, DMF.
A third strategy towards furanosyl α-amino acids has been reported by Dondoni et al. (Scheme 5) [25]. It involves the preparation of a thiazolyl ketol acetate, a key intermediate in the formylation of d-mannose at the anomeric position. Treatment of either α- or β-anomer 32 and 33, which are readily available through the addition of 2-lithiothiazole to the corresponding lactone 31 and subsequent acetylation, with trimethylsilyl triflate and trimethylsily azide, proceed stereopecifically giving rise, in both cases, to the azido-α-mannofuranose 34 in 84% yield. Cleavage of the thiazole ring of 34, followed by oxidation and esterification, afforded azido ester 36. The reduction of the azido group afforded in the early stages of the reaction exclusively the amino ester 37, but as the reaction proceeded, formation of the anomer 38 was also observed, resulting in a 1:5 mixture of 37 and 38 being isolated in quantitative yield. The existing equilibrium between 37 and 38, which is very likely to be facilitated by an α-imino ester intermediate, was demonstrated by the formation of anomeric mixtures of the same 1:5 ratio from either pure 37 or 38.
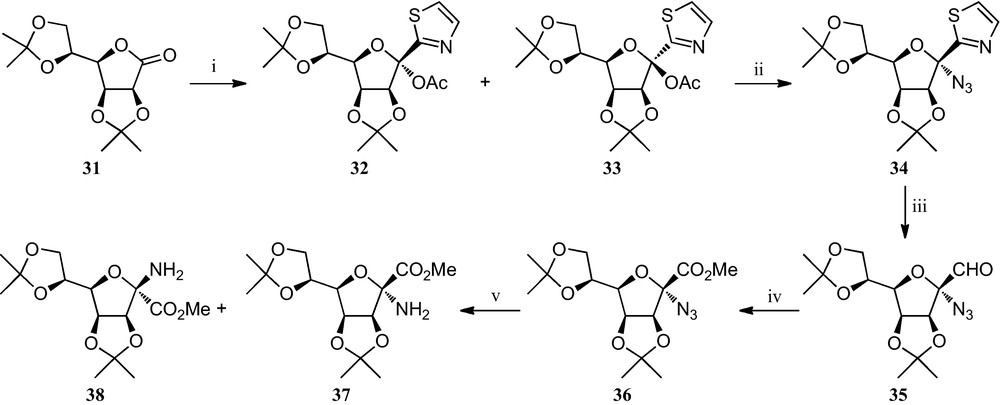
(i) [25a]; (ii) TMSOTf, TMSN3, CH2Cl2; (iii) (a) TfOMe, CH3CN; (b) NaBH4, MeOH; (c) HgCl2, H2O, CH3CN; (iv) (a) Ag2O, NaOH, H2O, THF; (b) CH2N2, MeOH, Et2O; (v) H2, Pd/C, t-BuOH, H2O.
2.2 Synthetic approaches towards pyranoid sugar α-amino acids
Some of the above strategies towards furanoid sugar α-amino acids have also been applied to the preparation of pyranoid sugar α-amino acids. Thus, Fleet et al. reported an approach of the synthesis of d-mannopyranose derivatives incorporating an anomeric amino acid moiety from the seven carbon lactone 39 (Scheme 6). It involved the preparation of an N-acylated bicyciclic[2.2.2]lactone 43, formed via an oxidative ring closure. This gyclopeptide analogue was easily transformed into mannopyranose derivative 44 containing an α-amino acid moiety at the anomeric position. Alternatively, derivative 43 could also be converted into pyranoid diketopiperazine 45, which on treatment with base afforded furanoid diketopiperazine 46 [26].
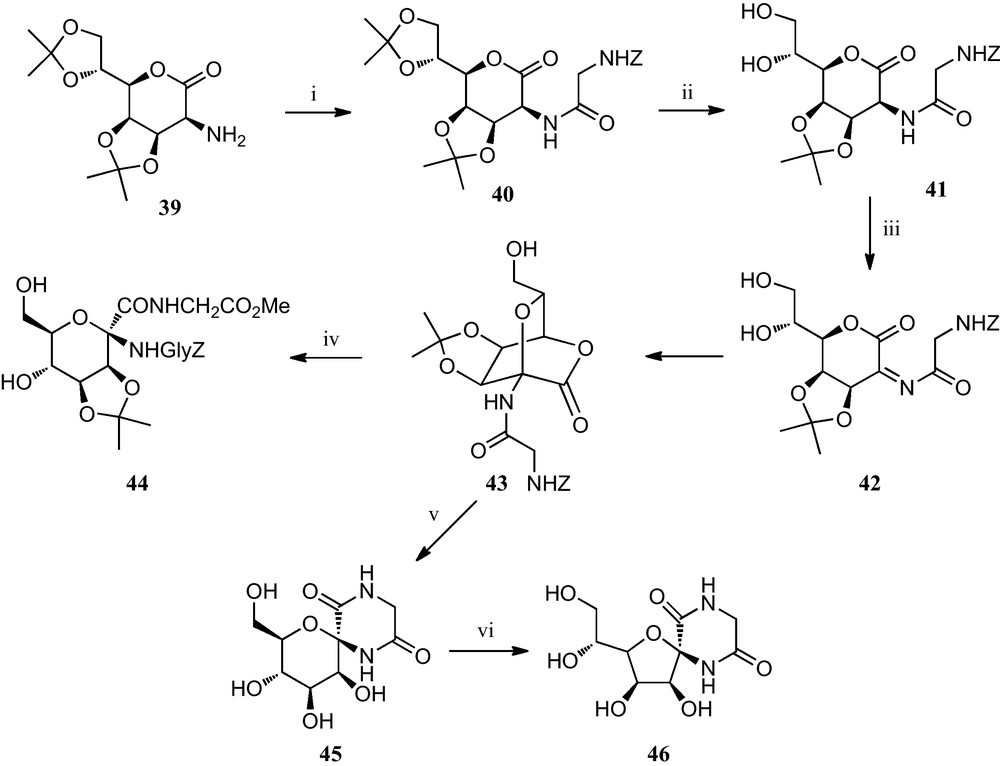
(i) Z-Gly-OH, ClCO2Et, Et3N, THF–MeCN (1:1) (ii) aq. AcOH (iii) N–bromophthalimide, NaOAc, CH3CN; (iv) H-Gly-OMe.HCl, NaOAc, DMF (v) (a) H2, Pd-black, EtOAc; (b) TFA/H2O (1:1); (vi) KOtBu, DMF.
Dondoni also applied his strategy for the preparation of furanoid sugar α-amino acids to the synthesis of pyranoid sugar α-amino acids, as exemplified in Scheme 7, where the transformation of 1,5-gluconolactone 47 into pyranoid SAA 52 is depicted [25,27].

(i) [25a]; (ii) (a) Ac2O, pyridine; (b) TMSOTf, TMSN3, CH2Cl2; (iii) (a) TfOMe, CH3CN; (b) NaBH4, MeOH; (c) HgCl2, H2O, CH3CN; (iv) (a) Ag2O, NaOH, H2O, THF; (b) CH2N2, MeOH, Et2O; (v) H2, Pd/C, t-BuOH, H2O.
3 Polyhydroxylated 1-aminocycloalkanecarboxylic acids
At present, a limited number of polyhydroxylated 1-aminocyclopentane and 1-aminoclohexanecarboxylic acids have been reported. All them belong to some one of the general structures displayed in Fig. 5.
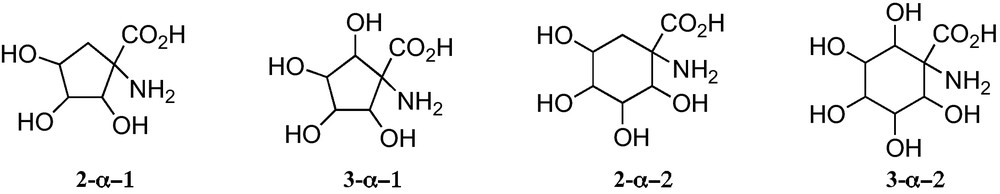
Trihydroxylated 1-aminocyclopentanecarboxylic acids 2-α−1 can be considered convenient mimetics of furanoid sugar α-amino acids 1-α (n = 1) (Fig. 1), that allowed one to avoid the anomeric configurational instability of the latter. In addition, 1-aminocyclopentanecarboxylic acids 2-α−1 and 3-α − 1 are closely related to the corresponding unsubstituted cyclopentane α-amino acids 4-α (Fig. 1), a family of amino acids that have been incorporated into peptides of chemical and biological interest [28]. These polyhydroxylated cyclopentane-based amino acids open paths for the generation of chemical diversity in this field and for the access to hydrophilic or hydrophobic amino acids, by protecting or deprotecting their hydroxy substituents. Similar properties should be expected for 1-aminocyclohexanecarboxylic acids 2-α−2 and 3-α−2.
3.1 Synthetic strategies towards polyhydroxylated 1-aminocyclopentane-carboxylic acids
At present only type 3-α−1 polyhydroxylated cyclopentane α-amino acids have been prepared from carbohydrates [29].
Fleet et al. reported the preparation of tetrahydroxylated cyclopentane α-amino acids 57a–f, which where efficiently prepared by means of short routes with control of the stereochemistry at all the five carbons bearing functional groups (Scheme 8) [29].
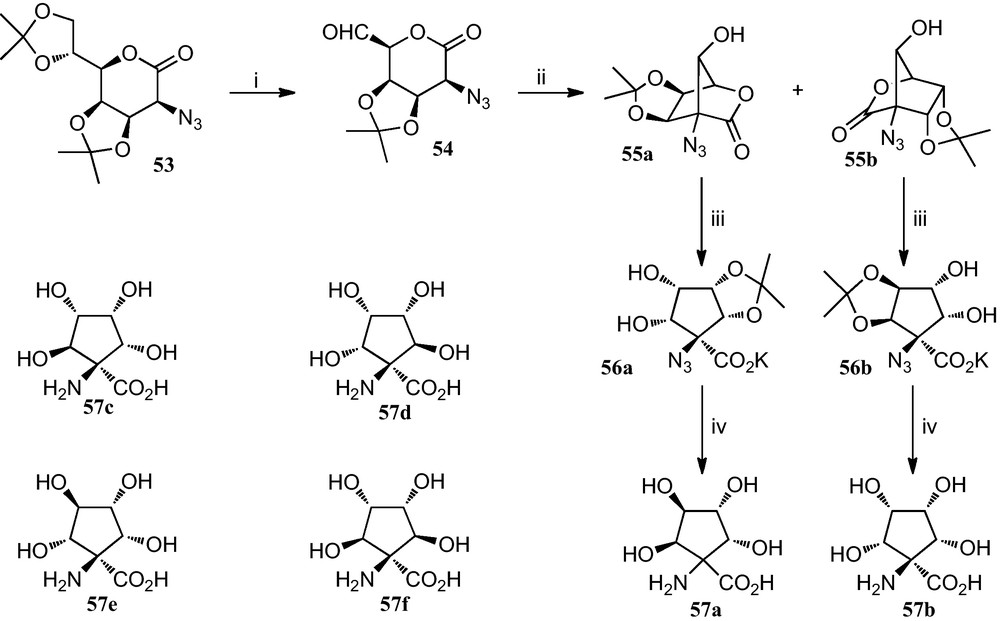
(i) MeCOOH, H2O; then HIO4 in THF (ii) KF or NaN3 in MeCN (iii) aq. base (iv) (a) H2, Pd-black, EtOH; (b) TFA, H2O.
For the synthesis of cyclopentane amino acid 57a, the diacetonide 53 was converted into the azidoaldehyde 54, which when reacted with potassium fluoride or sodium azide in acetonitrile afforded azidolactones 55a and 55b in a combined yield of 73% and a ratio 4:1. Reaction of 55a with aqueous potassium carbonate gave the carboxylate salt 56a, which was hydrogenated in the presence of palladium black to give an amine which afforded the amino acid 57a after removal of the protecting group. A similar reaction sequence provided amino acid 57b from 55b via the potassium salt 56b.
On the other hand, performing the deprotection and the hydrogenation prior to the opening of the lactone ring of lactones 55a and 55b allowed access to amino acids 57c–f.
3.2 Synthetic strategies towards polyhydroxylated 1-aminocyclohexane-carboxylic acids
A slight modification of the synthetic route towards polyhydroxylated cyclopentane α-amino acids stated in Scheme 8 allowed the preparation of tetrahydroxylated cyclohexane α-amino acid 61 [30]. Basic treatment of cyclic sulphate 58 induced a cyclization by attack of the carbanion at C-2 onto the primary carbon of the sulphate to afford bicyclic azidolactone 59. Removal of the isopropylidene and subsequent hydrogenation of the azido function gave bicycle 60, which upon treatment with aqueous triethylamine gave the cyclohexane α-amino acid 61 (Scheme 9).
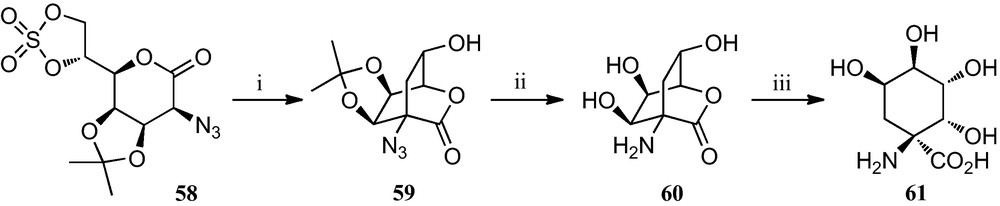
(i) KF, MeCN; (ii) (a) TFA, H2O; (b) H2, Pd-black, EtOH; (iii) aq. base.
A synthetic approach based on nitrosugars has been developed for the preparation of pentahydroxylated cyclohexane α-amino acids (Scheme 10).
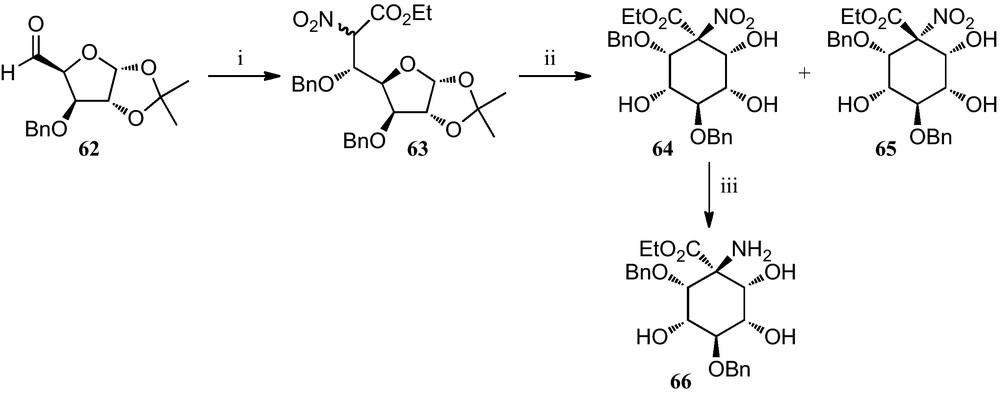
(i) (a) O2NCH2CO2Et, TBAF, ClTBDMS, Et3N, THF; (b) 2,2,2-benzyltrichloroacetimidate, TfOH, CH2Cl2, C6H12; (ii) (a) TFA, H2O; (b) 2% aq. HNaCO3, MeOH; (iii) H2, Raney-Ni, MeOH.
Reaction of the xylofuranose derivative 62 with ethyl nitroacetate provided an epimeric mixture of sugar α-nitro acetates 63, which, when subjected to a reaction protocol involving an intramolecular Henry reaction, provided the separable epimeric mixture of cyclohexanes 64 and 65. Compound 64 was converted into the corresponding amino acid ester 66 [31].
4 Furanoid and pyranoid sugar β-amino acids
The furanoid and pyranoid sugar β-amino acids here reported comprise only amino and carboxyl moieties which are both directly linked at contiguous positions on the corresponding rings. They can be classified according to the five types displayed in Fig. 6.
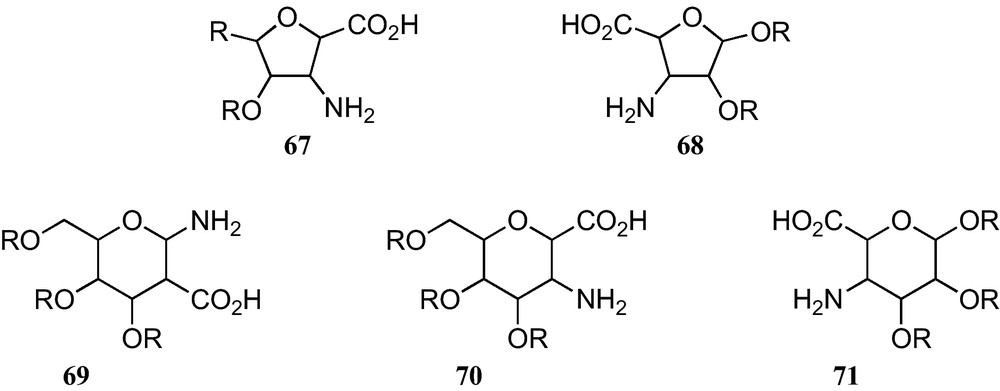
As amino acids 67 and 70 have either a carboxyl substituent at their anomeric position, they are anomerically stable, but amino acids 68, 69, and 71 require permanent protection of their anomeric amino or hydroxy substituents, in order to avoid the presence of anomeric mixtures.
The present interest into these five types of sugar β-amino acids is due to their structural relationship to the corresponding 2-aminocycloalkanecarboxylic acids, which, as conformationally constrained cyclic β-amino acids, have become in recent years a hot topic in synthetic and medicinal chemistry. In fact, these compounds are present in natural products, peptides, β-lactams and antibiotics. In addition to their pharmacological interest, these compounds have been used as chiral auxiliaries and as building blocks for the preparation of biologically active peptides and heterocyclic compounds (Section 5) [32].
All of these five types of β-amino acids have been prepared from sugars by means of the synthetic approaches stated in sections 4.1 and 4.2.
4.1 Synthetic approaches towards type 67 and type 68 furanoid sugar β-amino acids
Kessler et al. have demonstrated that incorporation of a SAA into a peptide sequence creates a conformationally rigid peptide-like structure, indicating that these derivatives can be utilized as peptidomimetics [15b,33]. They reported in 2001 the preparation of some somatostatin analogues containing the new furanoid SAA 75 (Scheme 11) as a key structure-inducing template designed to introduce the possibility of varying the functional groups on the carbohydrate moiety and thereby fine-tune pharmacokinetic properties [34]. The synthesis of this SAA 75 began from cheap, commercially available diacetone d-glucose 72, which was converted into the azide precursor 73 via its triflyl derivative [35]. This was followed by a quantitative and selective deprotection that provided an exocyclic diol, which upon oxidation with KMnO4 resulted in the formation of compound 74. Finally, in a one-pot reaction, azide 74 was reduced and simultaneously Fmoc-protected to yield the SAA 75 in a 44% overall yield.
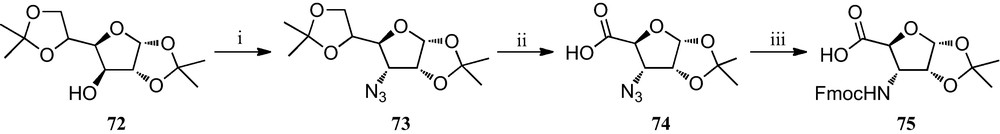
(i) (a) Tf2O, py, CH2Cl2; (b) NaN3, Bu4NCl (cat), DMF; (ii) (a) 77% HOAc; (b) NaIO4, MeOH, r.t.; (c) KMnO4, 50% HOAc, r.t.; (iii) H2, Pd/C, MeOH, Fmoc-Cl, NaHCO3, pH 8–9, THF, MeOH, r.t.
The preparation of the related methylamido amine has also been described [36].
Similarly, Fleet et al. prepared several 3-azidotetrahydrofuran-2-carboxylates from diacetone-d-glucose. The preparation of the 1S, 2R, 3S, 4R isomer 80 is depicted in Scheme 12; similarly, isomers 1S, 2R, 3R, 4R; 1S, 2S, 3R, 4R and 1S, 2S, 3R, 4S were also synthesized from diacetone-d-glucose [37].
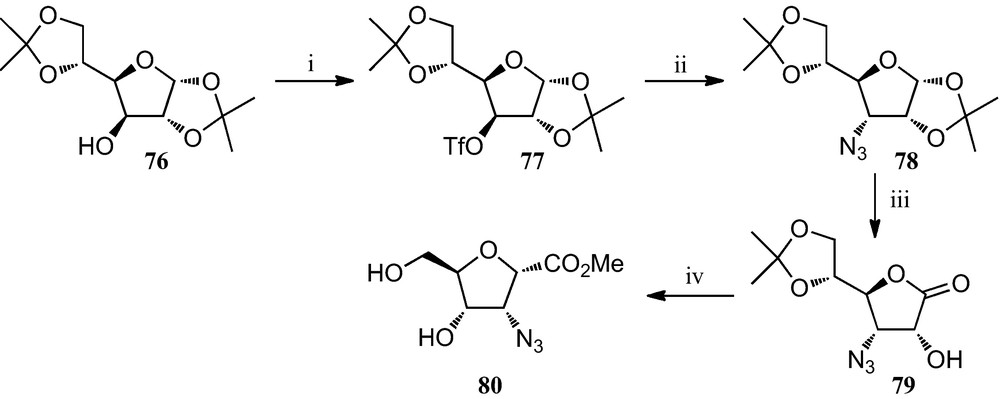
(i) Tf2O, pyridine, CH2Cl2; (ii) NaN3, DMF; (iii) (a) TFA/H2O; (b) Br2, BaCO3, dioxane, H2O; (iv) Tf2O, pyridine, CH2Cl2; then HCl, MeOH.
Additional work by Fleet et al. consisted of the preparation of several furanoid sugar β-amino acids by a different strategy involving the contraction of six membered sugar lactones to furanoid 2-carboxylates, where an azido group is introduced in the final step by nucleophilic displacement of a hydroxy group (Scheme 13). Thus, treatment of the O-triflyl derivative 82 of l-arabinose with methanolic hydrochloric acid promotes ring contraction affording methyl carboxylate 83, a process that was accompanied by the removal of the acetonide protecting group. This was followed by the conversion of this methoxylcarbonyl furane 83 into the corresponding isopropoxycarbonyl derivative 84, which when treated with ClTBDPS and imidazole resulted in the formation of a mixture of compounds 85 and 86. Finally, the free hydroxyl group in compound 86 was replaced by an azide via the corresponding triflate, to give azido ester 87, precursor of the corresponding amino acid [38].
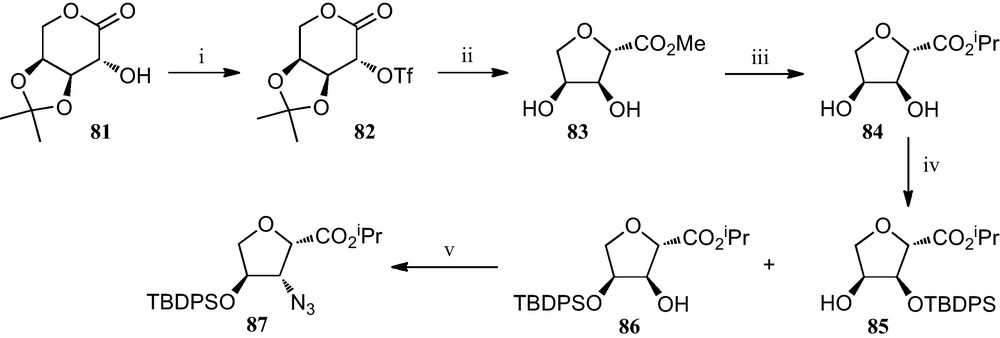
(i) Tf2O, py, CH2Cl2; (ii) 1% AcCl in MeOH; (iii) (a) 2N HCl; (b) 5% AcCl in i-PrOH; (iv) ClTBDPS, Im, DMF; (v) (a) Tf2O, py, CH2Cl2; (b) NaN3, DMF.
4.2 Synthetic approaches towards pyranoid sugar β-amino acids
It has been reported that the effect of glycosylation on bioactive peptides may be exploited to overcome shortcomings of potential peptide drugs [39]. In fact, it has been established that the attachment of saccharides to peptides improves bioavailability, increases resistance to proteases, improves water solubility, or enables the compounds to penetrate the blood–brain barrier. However, O- and N-linked glycopeptides are also subjected to both chemical and enzymatic degradation in vivo that limit their use as potential drugs. C-Glycosidic analogues possessing a C–C bond instead of a C–N or a C–O bond are thus valuable target molecules.
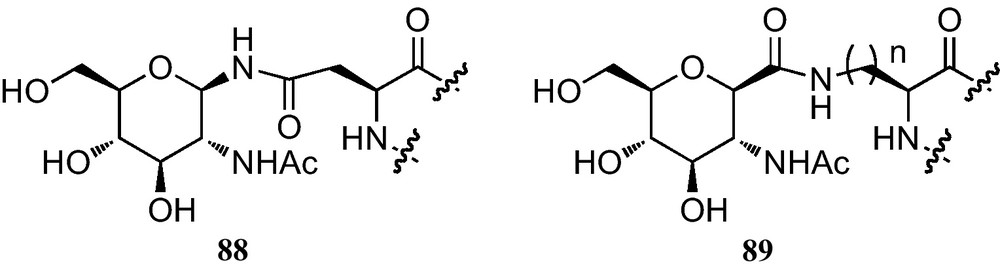
In the context of the preparation and incorporation of C-glycosylated amino acids into peptides, the synthesis of C-glycosidic analogues of N-4-(2-acetamido-2-deoxy-β-d-glucopyranosyl)-l-asparagine (89) containing a reversed amide bond as an isosteric replacement of the N-glycosidic linkage of 88 was reported (Fig. 7) [15b,34b,40].
The heptonic acid 93 required for the preparation of heptonamides 89 was obtained from 2-acetamido-3,4,6-tri-O-benzyl-2-deoxy-d-glucose 90, which in turn was synthesized in four steps from 2-acetamido-2-deoxy-d-glucose by a modified procedure of Warren et al. [41]. The synthesis of 91 was accomplished by addition of the α-configured sugar chloride obtained from 90 to a solution of Bu3SnLi at −78 °C (inverse addition) in 74% overall yield. Subsequent addition of carbon dioxide provided 93 in 83% yield, besides 16% of l-deoxysugar (Scheme 14).
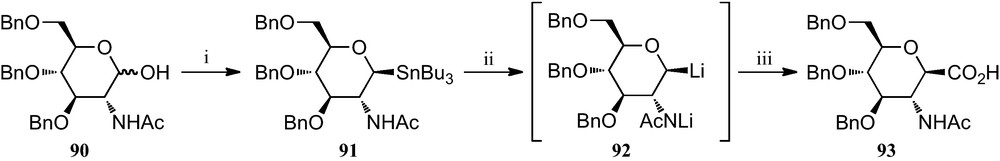
(i) (a) SOCl2; (b) Bu3SnLi; (ii) (a) n-BuLi; (iii) CO2.
A slight modification of this strategy, also reported in 1996, allowed the preparation of a carbopeptoid containing a C-1 carboxylate and C-2 amino group (Scheme 15) [42]. For the preparation of its monomeric component, d-glucosamine hydrochloride 98 was first converted into the known 1-cyano-2-phthalimido derivative 95, according to a published procedure [43]. The C-1 CN group of 95 was hydrated via treatment with 30% HBr-AcOH to furnish the CONH2 group 96 in 85% yield; this product was subsequently treated with Dowex 50W-X8 [H+] in refluxing MeOH to give the methyl ester derivative 97 in 97% yield. Removal of the phthaloyl group was accomplished by the successive treatment of 97 with aqueous LiOH and 3 N HCl, to give an amine derivative 98 as a HC1 salt, in 95% overall yield.
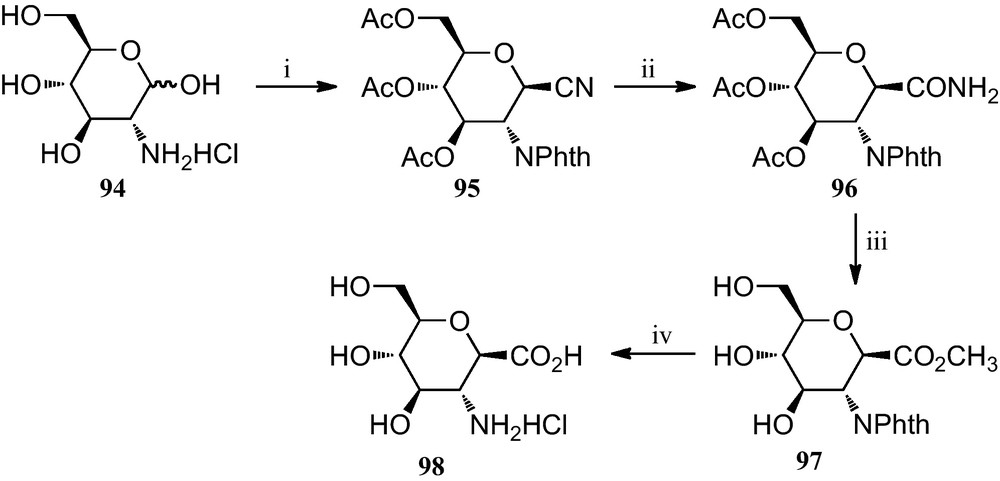
(i) [42a]; (ii) 30% HBr-AcOH; (iii) Dowex 50W-X8 [H+], MeOH; (iv) (a) six eq. of LiOH, MeOH/H2O (3:1); (b) 3 NHC1.
Vogel et al. reported the preparation of the novel 4-amino-5-carboxyl pyranosides 103 (Scheme 16) [44]. Treatment of 2,3-di-O-benzyl-6-O-(4-methoxybenzyl)-α-d-glucopyranoside 99 with methanesulfonyl chloride and subsequent substitution of the resulting 4-O-methanesulfonyl group with sodium azide gave, with inversion at C-4, the 4-azido derivative 100. After deprotecting at C-6 to give 101, the primary hydroxyl group was oxidized by means of the Corey procedure to a carboxyl group, and subsequently converted to a methyl ester by treatment with diazomethane, to afford 102. Conversion of this azidodeoxygalacturonate derivative 102 to the target amino ester 103 was achieved by reduction with hydrogen sulfide in pyridine.
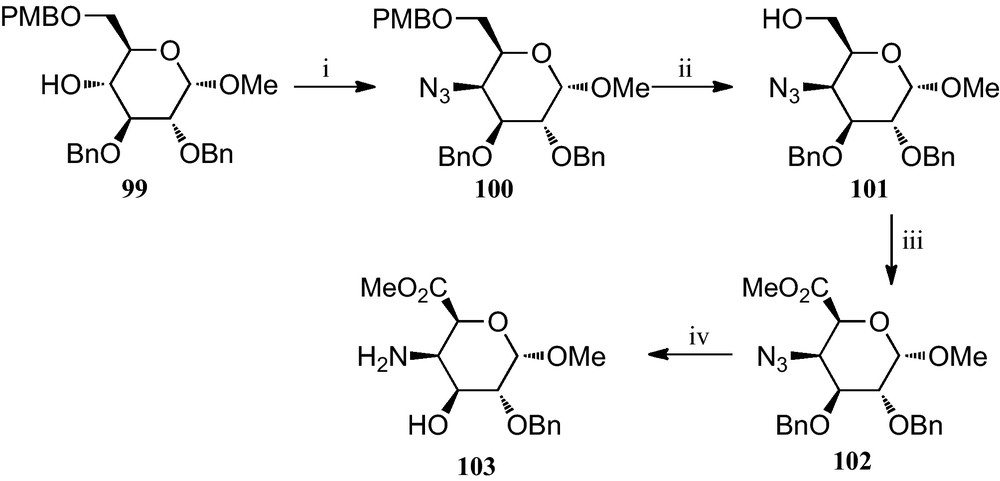
(i) (a) MsCl, Py; (b) NaN3, DMF; (ii) CAN, CH2Cl2; (iii) (a) PDC, DMF; (b) TMSCH2N2, MeCN; (iv) SH2, Py.
Preparation of 1-amino-2-acid pyranosides was also achieved using glycals as starting materials. Thus, glycal 104 was converted to the β-lactam 105, which was then acetylated to give 106. Opening of 106 with methanol finally gave amino ester 107 (Scheme 17) [45].
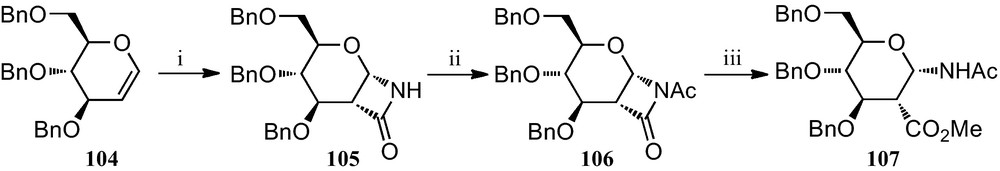
(i) (a) Cl3CC(=O)NCO, MeCN; (b) PhCH2NH2, MeCN; (ii) Ac2O, Et3N, CH2Cl2; (iii) MeOH.
5 Polyhydroxylated 2-aminocycloalkanecarboxylic acids
Alicyclic β-amino acids, which have received significant attention in recent years, include compounds of pharmacological interest, such as the naturally occurring β-amino acid (1R,2S)-2-aminocyclopentanecarboxylic acid (cispentacin) [46], an antifungal antibiotic, and (1R,2S)-2-amino-4-methylenecyclopentanecarboxylic acid (Icofungipen), a strong antifungal agent [47]. Derivatives of 2-aminocycloalkanecarboxylic acids can be used as chiral auxiliaries and have also served as important elements in the synthesis of β-lactams and heterocyclic compounds [48]. However, the main interest in these conformationally restricted β-amino acids arise from the ability of their oligomers to adopt well-defined secondary structures analogous to those of natural peptides, with their usefulness as building blocks for incorporation into peptides, in order to increase their metabolic stability and modify their biological activity [49].
The three types polyhydroxylated 2-aminocyclopentanecarboxylic acids and the two types of polyhydroxylated 2-aminocyclohexanecarboxylic acids at present reported are depicted in Fig. 8.
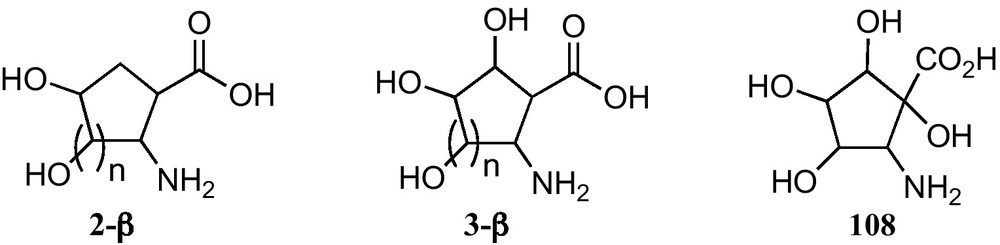
Given the biological importance of cyclic hydroxylated β-amino acids and their usefulness as suitable scaffolds for the preparation of rigid and water-soluble peptidomimetics, several procedures for their synthesis have been developed. However, much of the synthetic work in this field has focused on simpler cyclic β-amino acids and analogous studies of substituted derivatives are less well developed. In fact, the panel of known polyhydroxylated carbocyclic β-amino acids is limited, and this is probably due to the fact that only recently a few methods for their stereocontrolled preparation have been reported [50]. But their limited scope is the reason why preparation of these compounds remains a challenge for synthetic chemists [32e,39, 51].
At present, only polyhydroxylated cyclopentane β-amino acids 2-β, 3-β and 108 have been prepared from carbohydrates.
5.1 Synthetic approaches towards polyhydroxylated 2-aminocyclopentane-carboxylic acids
The first synthesis of a polyhydroxylated cyclopentyl β-amino acid is outlined in Scheme 18 [52]. An efficient aldol condensation of 2-iodo-5-formyl-1,5-lactone 109, catalyzed with potassium fluoride, gave bicyclic iodolactone 110, which upon treatment with potassium carbonate in methanol opened to furnish a cyclopentyl methyl ester that spontaneously underwent an intramolecular displacement of the iodine by the vicinal hydroxyl group to give epoxide 111. The opening of the epoxide ring by treatment with sodium azide, followed by acidic hydrolysis of the isopropylidene protecting group afforded azido ester 112, which upon hydrolysis of the methyl ester and reduction of the azido group should reveal the corresponding amino acid.
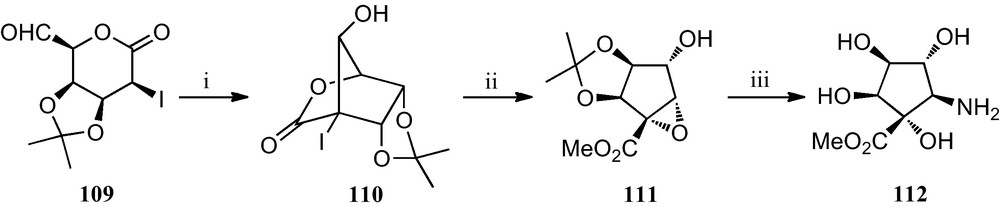
(i) KF, 18-crown-6, MeCN; (ii) K2CO3, MeOH; (iii) (a) NaN3, NH4Cl, aq. MeOH; (b) 40% aq. TFA; (c) H2, Pd-black, H2O.
More recently, several trihydroxylated 2-aminocyclopentanecarboxylic acids have been prepared by a novel intramolecular C-alkylation of nitro sugar nitronates (Scheme 19). Transformation of the nitrohexofuranose derivative 113 into the corresponding triflate 114, followed by a stereocontrolled cyclization of its nitronate 115, provided the bicyclolactone 116, which was reduced to amine 117 by catalytic hydrogenation. Finally, treatment of 117 with barium hydroxide resulted in the opening of its lactone ring, a process that allowed compound 118 to be obtained, which was the first reported trihydroxylated cis-2-aminocyclopentane carboxylic acid [53].
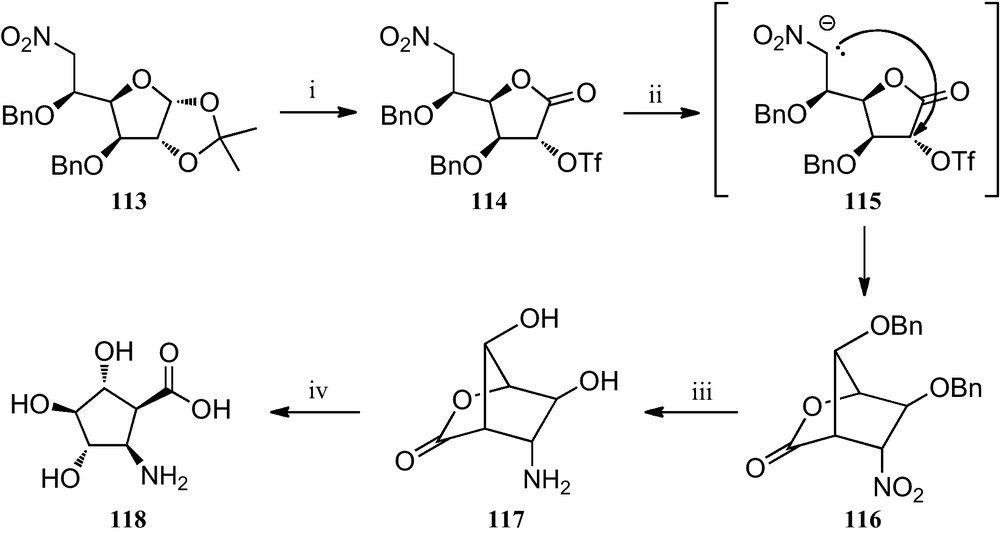
(i) (a) TFA/H2O (1:1); (b) Br2, BaCO3, dioxane/H2O (2:1); (c) Tf2O, pyridine, CH2Cl2; (ii) TBAF, THF; (iii) H2, Pd/C, dioxane/H2O/HCl; (iv) Ba(OH)2, H2O.
Similarly, when the 6-nitro-d-glucofuranose derivative 119 was subjected to a similar reaction protocol (Scheme 20), a stereocontrolled formation of the bicyclic nitrolactone 122 was observed, which upon treatment with sodium methoxide in dry methanol opened to furnish methyl 2-nitrocyclopentane 123. Finally, catalytic hydrogenation afforded compound 124, which formally constitutes the first described trihydroxylated trans-2-aminocyclopentane carboxylic acid [54].
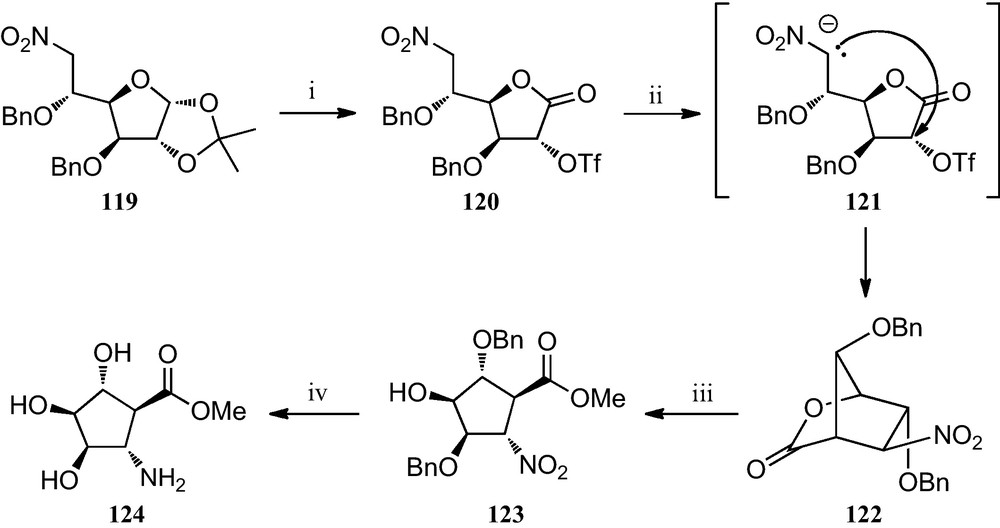
(i) (a) TFA/H2O (1:1); (b) Br2, BaCO3, dioxane/H2O (2:1); (c) Tf2O, pyridine, CH2Cl2; (ii) TBAF, THF; (iii) MeONa/MeOH (0.5 M); (iv) H2, Pd/C, MeOH, citric acid.
Moreover, a specific protocol has been developed for the incorporation of this amino acid derivative 124 into peptides (Scheme 21). It involves a selective reduction of the nitro group of bicyclic lactone 122 to furnish amino derivative 125, which has been used as the basis for the construction of peptides containing this β-amino acid. Freshly isolated 125 was subjected to peptide coupling with benzyloxycarbonylglycine, giving dipeptide 126, which was converted into acyl azide 128 via hydrazide 127. Finally, reaction of 128 with methoxycarbonylglycine provided tripeptide 129 [54].
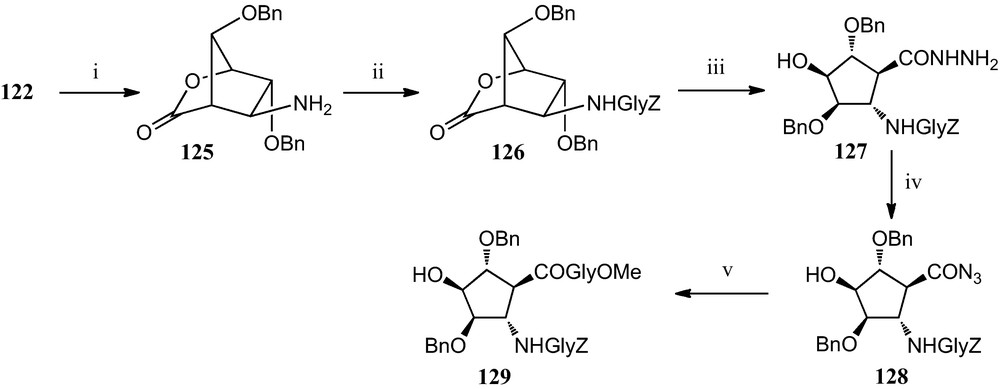
(i) H2, Raney-Ni, MeOH; (ii) ClCO2Et, Z-gly, Et3N; (iii) H2NNH2, MeOH; (iv) t–BuNO2, HCl, DMF, dioxan. (v) Gly-CO2Me, Et3N, DMF.
In a modification of this route, when the nitroglucofuranose derivative 119 (the starting material for route in Scheme 20) was now treated with acetyl chloride in methanol, it gave the epimeric mixture 130, which readily underwent the expected C-alkylation of the starting nitronates 132, that resulted in the formation of the anomeric mixture of bicylic compounds 133. Hydrolysis of this mixture, followed by immediate oxidation with sodium chlorite provided the desired 5-nitrocyclopentanecarboxylic acid 136a, which when reacted with trimethylsilyldiazomethane allowed methyl ester derivative 136b. Finally, catalytic hydrogenation of 136b provided amino acid 124 more efficiently [55] (Scheme 22).
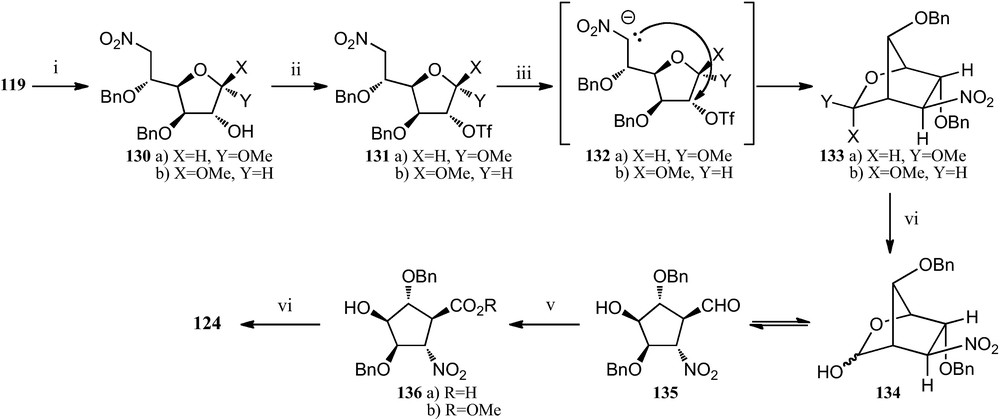
(i) AcCl, MeOH; (ii) Tf2O, pyridine, CH2Cl2; (iii) TBAF, THF; (iv) TFA/H2O (3:1); (v) NaClO2, NaH2PO4, 2-methyl-2-butene, t-BuOH/H2O (1:1); (vi) (a) TMSCHN2, Et2O/MeOH (7:2); (b) H2, Pd/C, MeOH, citric acid.
This efficient and elegant first reported strategy for the synthesis to tetrahydroxylated cyclopentyl β-amino acids has the evident limitation that it can provide access only to eight amino acids, i.e. just those resulting from the eight hexoses that satisfy the stereochemical requirements for the key intramolecular nitronate alkylation involved in their preparation (d-glucose, d-idose, d-alose, d-talose and their respective enantiomeric l-hexoses).
However, this strategy can be adapted to the preparation of additional members of this family of β-amino acids, as exemplified by the preparation of β-amino acid 141 (Scheme 23) Thus, after β-amino acid 138 was obtained from d-glucose following this nitrosugar-based strategy, it was readily converted into its epimer 140 via its O-trifyl derivative 139, according to the protocol depicted in Scheme 23. Finally, a catalytic hydrogenation provided the expected amino acid ester 141 [56].
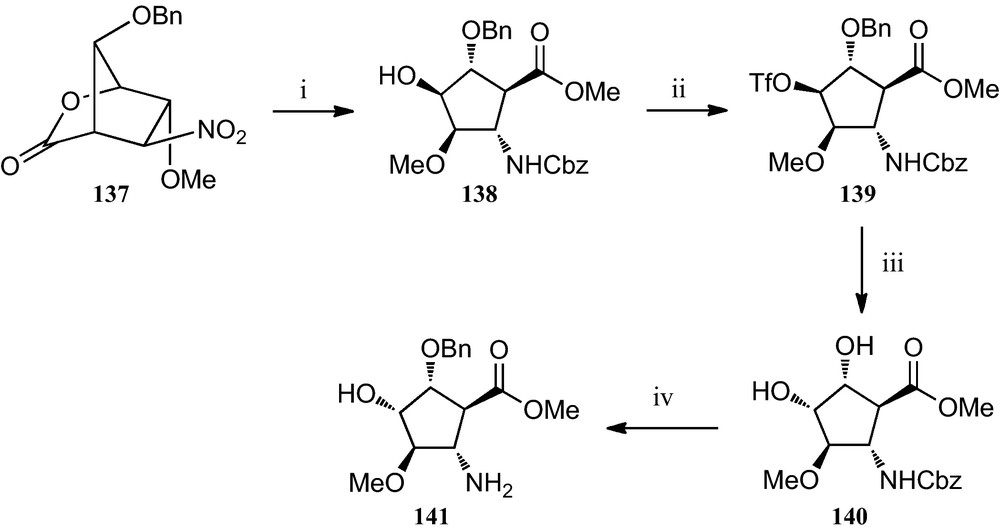
(i) AcCl, MeOH; (ii) Tf2O, pyridine, CH2Cl2; (iii) (a) CF2CO2Na, DMF; (b) MeONa, MeOH; (iv) H2, Pd/C, MeOH, citric acid.