

Apratoxin A and analogues are a recently discovered class of cyclodepsipeptides from marine origin, which possess high cytotoxicity (Fig. 1) [1–4]. Due to their potent antitumoral activity and uncommon mechanism of action, they have attracted much attention. Thus, total synthesis of apratoxins became such a competitive challenge for chemists that several total syntheses have been already reported [5–11], including solid supported synthesis of the oxazoline analogue that we have described [12]. Moreover, recently published biological studies prompted us to put on view our progress in the synthesis of new analogues [13–16].
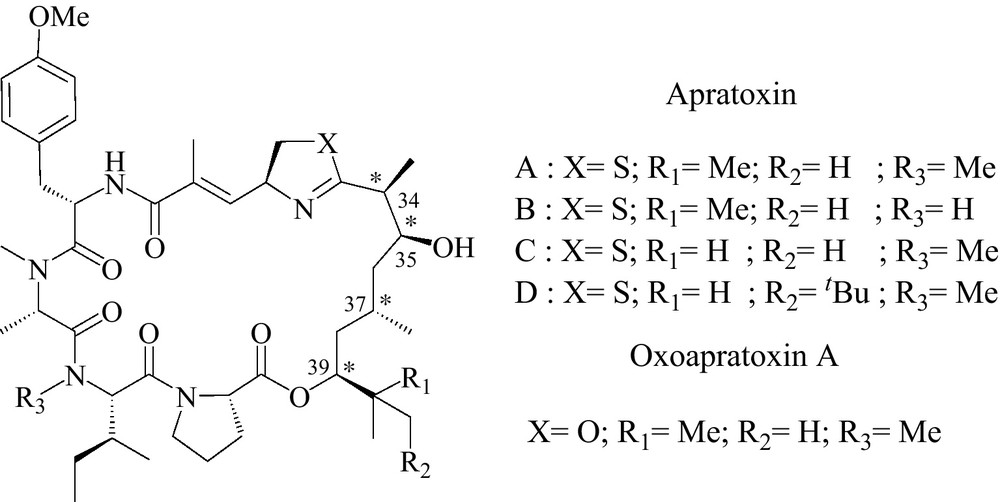
Apratoxin A and analogues.
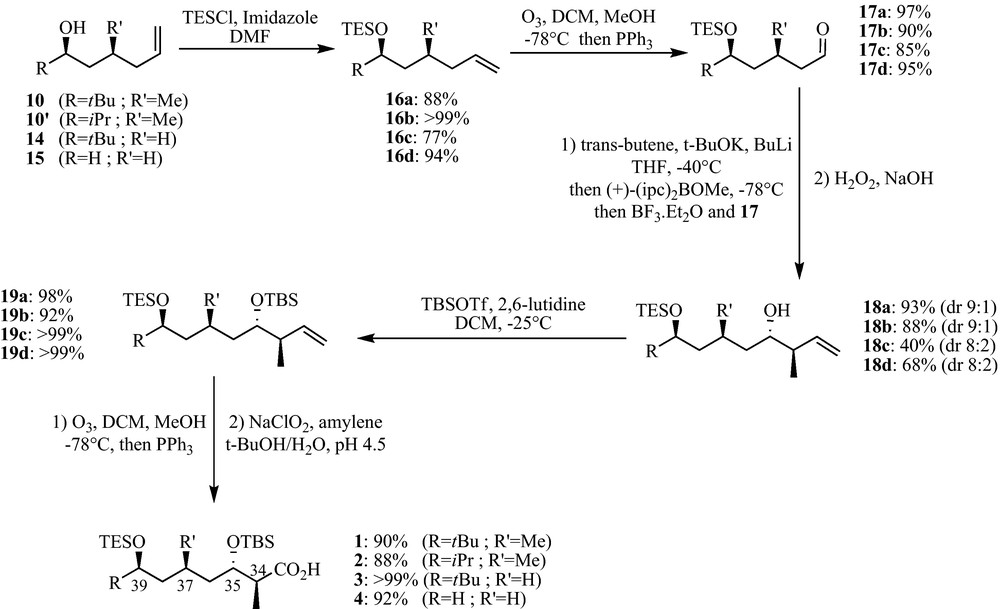
Indeed, towards our efforts to supply an easy and versatile synthesis of apratoxin analogues, we developed an efficient synthetic pathway to provide the polyketide moiety of apratoxin A and some of its analogues using a common key step. As a proof of efficiency, the polyketide fragment corresponding to apratoxin A has been synthesised in a gram scale. The four synthesised polyketides are displayed in Fig. 2.
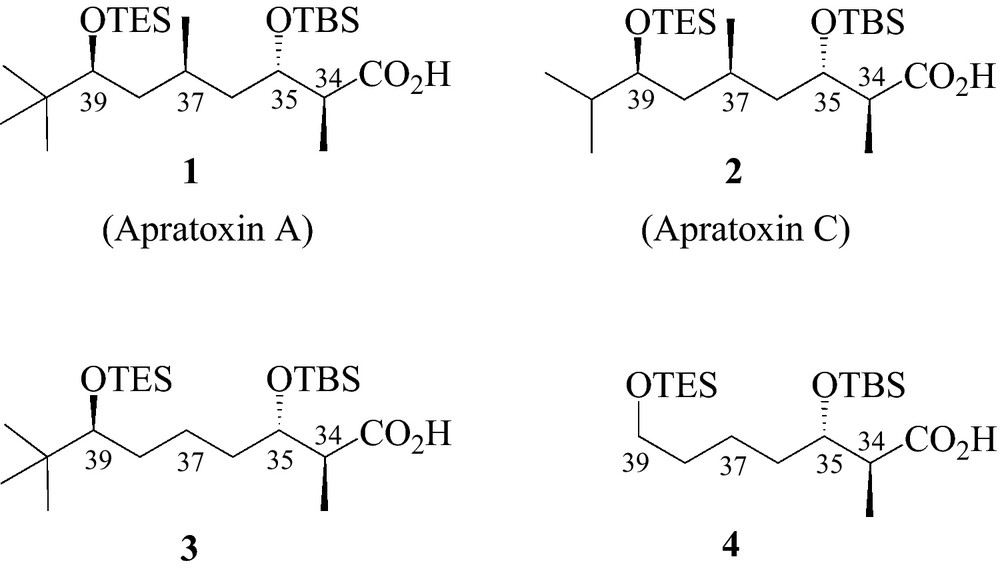
The four synthesised polyketides.
The polyketide fragments corresponding to apratoxin A (1) and apratoxin C (2) (Fig. 2) were first synthesised starting from pivalaldehyde 5 and butyraldehyde 5′ respectively, through a List's aldolisation reaction using proline as organocatalyst to give the corresponding hydroxyketones (Scheme 1) [17].
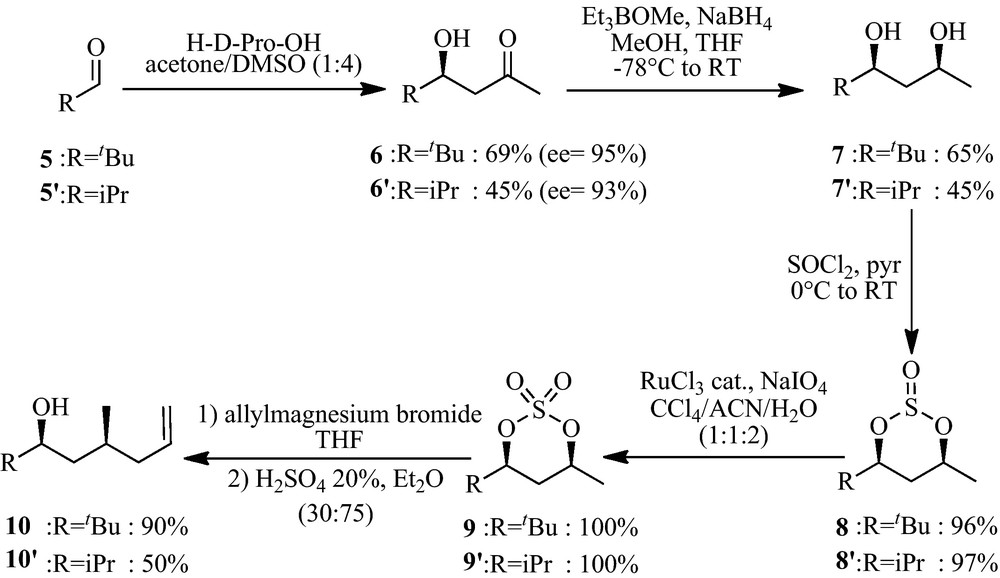
It is noteworthy that the butyraldehyde gave a lower yield, as already described by List. The diastereoselective reduction of the hydroxyketone 6 and 6′ under Prasad reaction conditions furnished the corresponding diols 7 and 7′, with a better diastereoselectivity for the pivalaldehyde (dr = 95:5 for 7, 65:35 for 7′), due to steric hindrance. In both cases, the diastereoisomers have been separated by silica gel chromatography (yields reported on Scheme 1 are given for the good diastereoisomer) [18]. The cyclic sulfate was obtained by cyclic sulfite formation (8 and 8′) with thionyl chloride followed by oxidation with RuCl3 and NaIO4 as cooxidant (9 and 9′) [19]. The sulfate was then subjected to a regioselective nucleophilic substitution with allylmagnesium bromide. When R = iPr (9′), the regioselectivity of the reaction was not complete due to less steric hindrance, as we observed the formation of its regioisomer in a 30% yield (50% yield reported on Scheme 1 is given for the good regioisomer). The substitution reaction induced complete inversion of configuration (SN2) in both cases. Final hydrolysis of the newly formed sulfate furnished the free alcohol (10 and 10′).
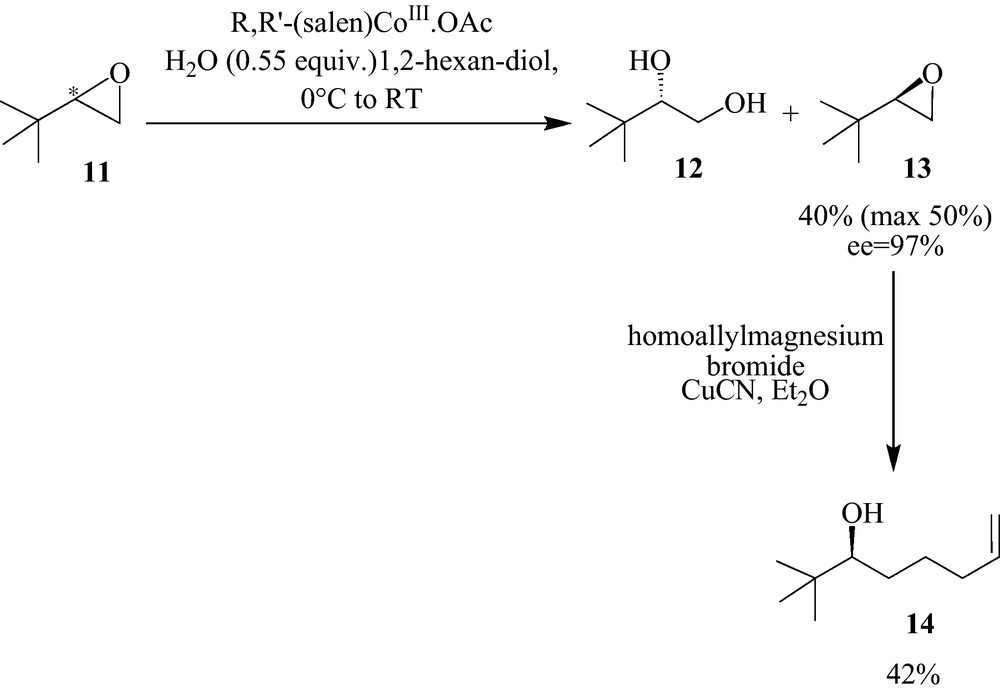
For the analogue 3 (Fig. 2), we started from the racemic epoxide 11. Hydrolytic kinetic resolution furnished the pure (R) epoxide 13 (ee = 97% obtained from optical rotation) [20], which was subjected to a regioselective nucleophilic substitution with homoallyl magnesium bromide to yield 14 (Scheme 2).
For the less hindered analogue 4 without alkyl group, we started the synthesis from the commercially available hex-5-en-1-ol (15).
From this stage, the rest of the synthesis was identical for all the four analogues and was performed as follows. The free alcohol was first silylated with TESCl (16), and the double bond was subjected to oxidative cleavage with ozone to give the corresponding aldehyde (17).
Then, our first trial to obtain the two last stereocenters was a non-Evans anti-aldolisation type reaction [21], but various attempts to furnish the aldol adduct were unsuccessful. Thus, we skipped to a Brown's crotylation reaction, which turned out to be very efficient to yield the corresponding homoallylic alcohol (18) [22].1
(+)-α-methoxydiisopinocampheyl borane was used as chiral auxiliary to obtain the desired enantioselectivity at the C35 chiral center, and the trans-butene was used as adduct to give the anti-compound with a good diastereoselectivity (dr = 8:2 to 9:1, determined from the NMR spectrum). We have been able to isolate the syn diastereoisomers by silica gel chromatography. It is noteworthy that the compounds 17c and 17d which did not possess a methyl group in the β position from the aldehyde have less diastereoselectivity, probably due to a loss a steric hindrance. The latter was then protected as TBDMS silyl ether (19), followed by ozonolysis to give an aldehyde (20), which was oxidized with NaClO2 into its corresponding carboxylic acid to furnish the four corresponding polyketide moities [23] (Scheme 3).
In summary, we described the syntheses of the polyketide moiety of apratoxins A and C and two other polyketide analogues. We stuck here to the stereochemistry of the natural compound, but there is the possibility to choose independently all the four chiral center's stereochemistry. Thus, we demonstrated the versatility of this synthesis, since there is also the possibility to introduce different alkyl groups at the C39 position, giving the potentiality to synthesise numbers of new polyketide analogues, and consequently, using our supported strategy, new apratoxin analogues [12]. The synthesis of some of these apratoxin analogues is already planned for future work, opening new perspective for future SAR studies.
Conflict of interest statement
There is no conflict of interest between the coauthors.
Acknowledgements
We thank La Ligue Contre le Cancer for financial support.
1 General procedure for the crotylation reaction: To an excess of trans-butene was added potassium t-butylate (2 equiv.) at −78 °C. After 10 min stirring at −78 °C, n-BuLi (2 equiv., 1.6 M/hexane) was added dropwise. The temperature was then allowed to raise to −45 °C and the mixture was stirred for 30 min. The mixture was then cooled down to −78 °C and a solution of diisopinocamphenylborane (2 equiv.) in a minimum of THF was added dropwise. After stirring for 45 minutes at −78 °C, BF3.Et2O (2.6 equiv.) was quickly added, immediately followed by a solution of aldehyde (1 equiv.) in THF. The mixture was stirred at −78 °C for 5 hours and quenched by addition of 3N NaOH and 30% H2O2. After 1 hour stirring at room temperature, the mixture was diluted with AcOEt and brine. The aqueous layer was extracted 3 times with AcOEt, and the combined organic layers were washed with brine, dried over Na2SO4 and concentrated under reduced pressure. Silica gel chromatography (hexane/AcOEt 93:7, Rf: ∼0.4) afforded the homoallylic alcohol as a colorless oil.