

1 Introduction
Les bislactones 1 se trouvent au cœur d’un certain nombre de produits naturels, dotés notamment d’activité biologique, et certaines d’entre elles sont reconnues en tant que modulateurs de fonctions du système nerveux central, notamment par inhibition de la catéchol-O-méthyltransférase [1]. Cependant elles sont essentiellement mentionnées en tant que précurseurs des furofuranes 2 (Schéma 1), composés classés dans la famille des lignanes [2]. Par voie de conséquence, si des efforts particuliers ont été consacrés au développement de méthodes synthétiques pour accéder aux furofuranes, peu de travaux ont été consacrés au sous-groupe de leurs précurseurs potentiels, les bislactones [2,3]. Les différents schémas réactionnels décrits dans la littérature sont principalement axés sur la synthèse de bislactones symétriques 1 comportant deux unités aryliques identiques (Schéma 1, Ar1 = Ar2) sur les positions 3 et 6. Les bislactones sont alors obtenues par diverses dimérisations oxydatives de dérivés de l’acide cinnamique [4]. Néanmoins, un certain nombre de travaux orientés vers la synthèse de dérivés dissymétriques (Schéma 1, 1, Ar1 ≠ Ar2) proposent une construction s’appuyant sur des dérivés de l’anhydride maléique en tant qu’intermédiaires clés [5].

L’intérêt lié à la synthèse du (±)–gmelinol a conduit quelques équipes à envisager la synthèse de composés du type 3 (Schéma 2) comportant trois substituants dont un (R3 = SMe, OH, Me) à la jonction des cycles en supplément des deux groupes aryles caractéristiques des lignanes (R1 et R2). La première unité lactonique peut être construite à partir du bisanion dérivant d’un 1,4-diester. Elle conserve alors une fonction ester qui peut ensuite être engagée dans la cyclisation du second cycle lactone par une réaction de transestérification soit à l’aide d’un alcool généré par addition nucléophile sur un aldéhyde et traitement au TFA [6] soit par réduction d’une fonction cétone préexistante et cyclisation acido-catalysée [7]. Enfin un exemple permettant l’accès à des bislactones indifféremment substituées (R1, R2, R3) par des groupes aryles ou alkyles repose sur l’addition, avec réarrangement, d’aldéhydes sur les énolates de β-lactones substituées par un ester [8]. En règle générale, l’obtention des bislactones, ainsi que leur structure, est fortement conditionnée par la stéréochimie des composés de départ.
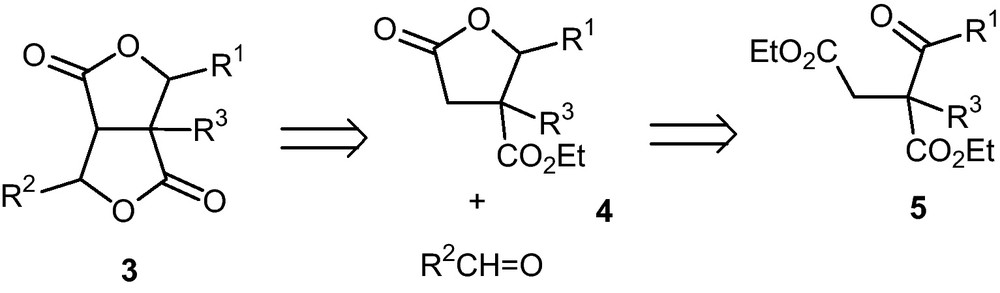
Nous avons récemment montré l’intérêt des lactones de type 4 en tant que base de départ pour la synthèse d’α-méthylène-γ-butyrolactones [9]. En prolongement de cette étude, nous avons envisagé une adaptation de la stratégie proposée comme moyen d’accès aisé et efficace aux bislactones 3 avec une grande latitude quant à la nature des substituants R1, R2, R3 (Schéma rétrosynthétique 2). Les lactones 4 resteraient construites par transestérification de l’un des esters du cétodiester de diéthyle 5 par un alcoolate généré par réduction du carbonyle [10]. La présence d’un centre carboné quaternaire en β sur 4 limitant la déprotonation à la position α, la seconde cyclisation pourrait se réaliser également par transestérification intramoléculaire directe de l’ester résiduel à l’aide de l’alcoolate intermédiaire résultant d’une addition nucléophile d’un aldéhyde sur cette position.
2 Résultats et discussion
La préparation préalable des cétodiesters parents 5a–c qui incorporent deux des substituants choisis pour les systèmes ciblés (R1 et R3) est réalisée à partir des β-cétoesters 6 et 7 selon une double séquence déprotonation/substitution par le bromoacétate d’éthyle, puis MeI ou BnBr. La réduction de la fonction cétone par NaBH4 dans EtOH se traduit par la formation concomitante des lactones 4a–c (Schéma 3) [9,10]. Il est intéressant de noter que le remplacement de l’ester benzylique par son équivalent éthylique modifie la stéréosélectivité de la réaction de cyclisation. Alors que la présence d’un ester benzylique orientait la synthèse vers la formation exclusive du diastéréoisomère cis [9], ces essais, conduits dans les mêmes conditions, nous ont fourni les lactones désirées 4a–c sous forme d’un mélange des deux diastéréoisomères cis et trans, séparables, de composition variable mais dans lequel aucune des deux formes n’est réellement prépondérante (Schéma 3, Tableau 1).

Rendements obtenus lors de la synthèse des lactones 4 et des bislactones 3.
| R1 | R3 | 4 | 3 | |||||
| Rdt (%) | cis/transa | R2 | Rdt (%) | cis/transb | ||||
| a | Ph | Me | 69 | 71/29 | aa | Et | 80 | 83/17 |
| ab | C6H13 | 78 | 85/15 | |||||
| ac | C8H17 | 70 | 73/27 | |||||
| ad | (p)MeO-C6H4 | 83 | cis | |||||
| b | Ph | Bn | 61 | 59/41 | ba | Et | 73 | cis |
| bb | C6H13 | 78 | cis | |||||
| bc | C8H17 | 65 | cis | |||||
| bd | (p)MeO-C6H4 | 42 | cis | |||||
| c | Me | Bn | 63 | 40/60 | cc | C8H17 | 75 | cis |
| ce | 2,3,4-(MeO)3-C6H2 | 80 | cis |
a Cis fait référence à (2S*,3S*), trans à (2S*,3R*).
b Cis fait référence à (3aR*,6R*), trans à (3aR*,6S*).
L’addition des différents aldéhydes a été réalisée sur l’énolate dérivé de chacun des deux diastéréoisomères obtenus lors de la formation des γ-butyrolactones 4a–c et généré par traitement au LHMDS à basse température. Les résultats obtenus diffèrent selon le diastéréoisomère utilisé. L’addition d’un aldéhyde sur l’anion des diastéréoisomères trans–4 (le groupement R1 se trouvant du côté opposé à la fonction ester) conduit à l’obtention d’une bislactone alors que la réaction d’un aldéhyde avec le diastéréoisomère cis–4 aboutit à la formation de l’alcool ouvert (Schéma 3, Tableau 1). Il est probable que le système anionique formé lors de l’étape de déprotonation attaque l’aldéhyde de façon à ce que l’alcoolate 9 ainsi généré se trouve du côté le moins encombré de la lactone. Dans le cas des diastéréoisomères trans–4, cette fonction alcoolate se trouve alors du même côté que la fonction ester permettant ainsi la cyclisation sous forme de bislactone.
L’alcool 8 obtenu lors de l’addition du para-anisaldéhyde sur le diastéréoisomère cis–4a a été soumis à une réaction de transestérification acido catalysée (APTS) à reflux dans le toluène. Si cette technique s’était révélée fructueuse dans le cas d’une lactone non substituée sur le carbone portant l’ester [6], malheureusement, même à une température plus élevée (reflux de toluène vs température ambiante), le composé s’est révélé réfractaire à ce processus de cyclisation, aucune bislactone n’étant obtenue dans ces conditions.
Si l’on excepte le cas de la réaction avec le para-anisaldéhyde, les bislactones 3aa–ac comportant un groupe R3 = Me ont été obtenues sous forme d’un mélange de deux diastéréoisomères dans lequel le diastéréoisomère (3a,6)-cis est largement prédominant. Pour les bislactones possédant un groupement R3 = Bn (3ba–bd et 3cc, 3ce), ce diastéréoisomère (3a,6)-cis est obtenu exclusivement. La stéréochimie des divers composés cycliques synthétisés a pu être déterminée à partir de l’analyse par diffraction des rayons X de 3ad qui montre sans ambiguïté une configuration (3a,6)-cis (Fig. 1).

Représentation ORTEP de la structure cristalline de 3ad. Sélection de données cristallographiques : C20H18O5 ; M = 338,34 g.mol−1 ; F(000) = 712 ; cristaux incolores ; Dx = 1,314 g cm−3 ; μ(Mo Kα) = 0,094 ; monoclinique P2(1)/c ; a = 10,225(2) Å ; b = 6,3779(13) Å ; c = 26,233(5) Å ; α = 90,00 ; β = 90, 948(4) ; γ = 90,00 ; V = 1710,5(6) Å3 ; Z = 4 ; T = 293(2) K.
La synthèse de bislactones présentant un groupement méthylène non substitué en position 6 a été envisagée initialement par addition de paraformaldéhyde selon la méthode employée précédemment pour la formation des bislactones 3. Cette voie de synthèse ne donnant que de très mauvais rendements en raison, vraisemblablement, de la nature hétérogène du milieu, une méthode alternative d’introduction de la fonction alcool primaire a du être envisagée. La stratégie choisie consiste en l’introduction préalable de la fonction alcool sous une forme masquée par substitution de l’anion dérivant d’une lactone 4. Puis, après déprotection, l’alcool primaire ainsi généré devrait permettre la cyclisation par transestérification acido catalysée [6]. Notre choix quant à l’électrophile s’est porté sur l’éther iodométhylbenzylique, généré in situ [11], pour sa réactivité ainsi que pour la facilité de la déprotection du groupe benzyle. Cette technique a donc été mise en œuvre sur les lactones trans–4a et cis–4a pour délivrer les composés désirés trans–10a et cis–10a avec de bons rendements (Schéma 4). La déprotection de l’alcool par hydrogénolyse catalysée au Pd/C donne accès aux alcools trans–11a et cis–11a. Le traitement dans le toluène à reflux en présence d’une quantité catalytique d’APTS conduit à la bislactone 3af dans le cas du composé trans–11a alors que le dérivé cis–11a conduit à l’α-méthylène-γ-butyrolactone 12 par déshydratation (Schéma 4).

3 Conclusion
Les butyrolactones polysubstituées, généralement obtenues sous forme de deux diastéréoisomères séparables lors de l’étape de purification, se sont avérées être une plate-forme intéressante pour la synthèse d’une variété de systèmes mono- ou bicycliques. En particulier le groupement fonctionnel de type ester positionné en β sur le cycle lactone peut se prêter à différentes transformations chimiques. La présence d’un groupement méthylène libre et déprotonable en position α sur le cycle lactone rend possible, par addition d’un aldéhyde sur l’énolate, l’échafaudage direct de systèmes bicycliques fusionnés, de type bislactone, et polysubstitués de manière dissymétrique. Dans le cas où une addition de l’aldéhyde se révèle peu rentable, l’introduction d’une fonction alcool masquée peut palier cette carence mais la cyclisation en bislactone nécessite une étape supplémentaire de transestérification acido-catalysée. Dans tous les cas les contraintes stériques liées à la présence des substituants sur le premier système lactonique imposent la présence du groupe carboxylate sur la face la moins encombrée du cycle condition pour assurer la fermeture ultime du second cycle lactonique.
4 Partie expérimentale
4.1 Généralités
La silice MERCK, Geduran SI 60, 0,040–0,063 mm, a été utilisée pour les chromatographies sur colonne sous pression (chromatographie « éclair »). Le tétrahydrofurane (THF) préséché sur Na2SO4 a été distillé sur sodium/benzophénone et sous atmosphère inerte (Ar) avant utilisation. Le toluène a été distillé sur hydrure de calcium Les spectres de RMN du proton, du carbone ont été enregistrés sur un appareil Bruker AM 300 WB (à 300 et 75 MHz respectivement) sur des solutions dans CDCl3 utilisant le TMS comme référence interne. Les analyses élémentaires ont été réalisées à l’aide d’un appareil Carlo Erba CHNS 11110. Pour l’analyse par diffraction des rayons X, les intensités diffractées ont été collectées à 100 K à l’aide d’un diffractomètre trois cercles Bruker AXS SMART équipé d’un détecteur bidimensionnel CCD avec la radiation Kα du molybdène (λ = 0,71073 Å). Les collectes ont été réalisées en balayages ω et φ. La structure a été résolue par méthodes directes et cartes de série de transformée de Fournier. La suite logicielle cristallographique SHELXTL a été utilisée pour tous les calculs [12].
4.2 Synthèse des cétodiesters 5a–c
Les β-cétodiesters 5a–c ont été préparés en suivant la méthode développée dans la littérature [9,10] au départ de benzoylacétate d’éthyle (6) ou d’acétoacétate d’éthyle (7).
4.2.1 2-Benzoyl-2-méthylsuccinate de diéthyle (5a)
Huile (50 % sur deux étapes). RMN 1H, δ (ppm) : 1,12 (t, 3 H, J = 7,1 Hz), 1,22 (t, 3 H, J = 7,1 Hz), 1,67 (s, 3 H), 3,06 (s, 2 H), 4,10 (q, 2 H, J = 7,1 Hz), 4,17 (q, 2 H, J = 7,1 Hz), 7,39–7,44 (m, 2 H), 7,50–7,55 (m, 1 H), 7,81–7,84 (m, 2 H). RMN 13C, δ (ppm) : 13,7 (CH3), 14,0 (CH3), 21,2 (CH3), 41,6 (CH2), 55,8 (C), 60,7 (CH2), 61,8 (CH2), 128,4 (CH), 128,5 (CH), 132,5 (CH), 135,7 (C), 170,5 (2 × CO), 196,6 (CO). Analyse, calculé pour C16H20O5 : C 65,74 ; H 6,90 ; trouvé : C 65,48 ; H 7,02 %.
4.2.2 2-Benzoyl-2-benzylsuccinate de diéthyle (5b)
F = 74–75 °C (38 % sur deux étapes). RMN 1H, δ (ppm) : 1,12 (t, 3 H, J = 7,1 Hz), 1,20 (t, 3 H, J = 7,1 Hz), 3,01 (s, 2 H), 3,49 (d, 1 H, J = 14,2 Hz), 3,60 (d, 1 H, J = 14,2 Hz), 4,09 (q, 2 H, J = 7,1 Hz), 4,16 (q, 2 H, J = 7,1 Hz), 6,99–7,02 (m, 2 H), 7,20–7,28 (m, 3 H), 7,40–7,45 (m, 2 H), 7,52 (t, 1 H, J = 7,3 Hz), 7,84 (d, 2 H, J = 7,3 Hz). RMN 13C, δ (ppm) : 13,7 (CH3), 14,1 (CH3), 37,9 (CH2), 39,2 (CH2), 60,6 (CH2), 60,8 (CH2), 61,9 (C), 127,1 (CH), 128,3 (CH), 128,4 (CH), 128,5 (CH), 130,3 (CH), 132,5 (CH), 135,6 (C), 136,6 (C), 170,6 (CO), 171,5 (CO) 195,9 (CO). Analyse, calculé pour C22H24O5 : C 71,72 ; H 6,57 ; trouvé : C 71,99 ; H 6,48 %.
4.2.3 2-Acétyl-2-benzylsuccinate de diéthyle (5c)
Huile (45 % sur deux étapes) [13].
4.3 Synthèse des lactones 4a–c. Procédure générale
Les lactones 4a–c ont été préparées par réduction/cyclisation à partir des β-cétodiesters 5a–c selon la méthode décrite dans un article précédent [9]. Le brut est fractionné par chromatographie sur gel de silice (acétate d’éthyle/éther de pétrole, 10/90). Les diastéréoisomères de la lactone 4c n’ont pu être parfaitement isolés mais le dérivé trans–4c a été obtenu dans un état de pureté suffisant pour être engagé dans l’étape suivante.
4.3.1 (2S*,3R*)-3-Méthyl-5-oxo-2-phényltétrahydrofuran-3-carboxylate d’éthyle (trans–4a)
Huile. RMN 1H, δ (ppm) : 0,97 (s, 3 H), 1,32 (t, 3 H, J = 7,1 Hz), 2,58 (d, 1 H, J = 17,1 Hz), 3,25 (d, 1 H, J = 17,1 Hz), 4,27 (q, 2 H, J = 7,1 Hz), 5,88 (s, 1 H), 7,26–7,29 (m, 2 H), 7,34–7,40 (m, 3 H). RMN 13C, δ (ppm) : 14,1 (CH3), 18,8 (CH3), 41,2 (CH2), 50,6 (C), 62,0 (CH2), 84,8 (CH), 125,8 (CH), 128,4, (CH), 128,6 (CH), 134,7 (C), 173,2 (CO), 174,1 (CO). Analyse, calculé pour C14H16O4 : C 67,73 ; H 6,50 ; trouvé : C 67,56 ; H 6,79 %.
4.3.2 (2S*,3S*)-3-Méthyl-5-oxo-2-phényltétrahydrofuran-3-carboxylate d’éthyle (cis–4a)
Huile. RMN 1H, δ (ppm) : 0,92 (t, 3 H, J = 7,1 Hz), 1,58 (s, 3 H), 2,55 (d, 1 H, J = 17,3 Hz), 3,21 (d, 1 H, J = 17,3 Hz), 3,65–3,82 (m, 2 H), 5,27 (s, 1 H), 7,25–7,28 (m, 2 H), 7,33–7,38 (m, 3 H). RMN 13C, δ (ppm) : 13,5 (CH3), 21,8 (CH3), 39,9 (CH2), 52,3 (C), 61,4 (CH2), 87,9 (CH), 125,6 (CH), 128,3 (CH), 128,9 (CH), 134,8 (C), 171,6 (CO), 174,7 (CO). Analyse, calculé pour C14H16O4 : C 67,73 ; H 6,50 ; trouvé : C 67,76 ; H 6,61 %.
4.3.3 (2S*,3R*)-3-Benzyl-5-oxo-2-phényltétrahydrofuran-3-carboxylate d’éthyle (trans–4b)
Huile. RMN 1H, δ (ppm) : 1,26 (t, 3 H, J = 7,1 Hz), 2,18 (d, 1 H, J = 13,7 Hz), 2,71 (d, 1 H, J = 17,7 Hz), 2,86 (d, 1 H, J = 13,7 Hz), 3,13 (d, 1 H, J = 17,7 Hz), 4,22 (q, 2 H, J = 7,1 Hz), 5,74 (s, 1 H), 6,96 (d, 2 H, J = 4,9 Hz), 7,16–7,29 (m, 3 H), 7,34–7,44 (m, 5 H). RMN 13C, δ (ppm) : 14,0 (CH3), 35,6 (CH2), 38,9 (CH2), 56,1, (C), 62,2 (CH2), 85,8 (CH), 126,5 (CH), 127,2 (CH), 128,5 (CH), 128,6 (CH), 128,8 (CH), 129,6 (CH), 134,4 (C), 137,7 (C), 172,5 (CO), 174,6 (CO). Analyse, calculé pour C20H20O4 : C 74,06 ; H 6,21 ; trouvé : C 73,84 ; H 6,48 %.
4.3.4 (2S*,3S*)-3-Benzyl-5-oxo-2-phényltétrahydrofuran-3-carboxylate d’éthyle (cis–4b)
Huile. RMN 1H, δ (ppm) : 0,92 (t, 3 H, J = 7,1 Hz), 2,70 (d, 1 H, J = 17,5 Hz), 2,96 (d, 1 H, J = 13,7 Hz), 3,07 (d, 1 H, J = 17,5 Hz), 3,61–3,71 (m, 2 H), 3,75–3,86 (m, 1 H), 5,43 (s, 1 H), 7,11–7,17 (m, 2 H), 7,27–7,38 (m, 8 H). RMN 13C, δ (ppm) : 13,6 (CH3), 35,9 (CH2), 40,9 (CH2), 61,5 (CH2), 57,9 (C), 87,2 (CH), 126,1 (CH), 127,4 (CH), 128,5 (CH), 128,7 (CH), 129,1 (CH), 129,9 (CH), 135,0 (C), 135,7 (C), 170,5 (CO), 174,9 (CO). Analyse, calculé pour C20H20O4 : C 74,06 ; H 6,21 ; trouvé : C 73,97 ; H 6,40 %.
4.3.5 (2S*,3R*)-3-Benzyl-2-méthyl-5-oxotétrahydrofuran-3-carboxylate d’éthyle (trans–4c)
Huile. RMN 1H, δ (ppm) : 1,21 (t, 3 H, J = 7,1 Hz), 1,53 (d, 3 H, J = 6,6 Hz), 2,57 (d, 1 H, J = 17,8 Hz), 2,63 (d, 1 H, J = 13,7 Hz), 2,97 (d, 1 H, J = 17,8 Hz), 3,31 (d, 1 H, J = 13,6 Hz), 4,14–4,20 (m, 2 H), 4,69 (q, 1 H, J = 6,6 Hz), 7,00–7,12 (m, 2 H), 7,20–7,29 (m, 3 H). RMN 13C, δ (ppm) : 14,1 (CH3), 15,0 (CH3), 35,5 (CH2), 36,4 (CH2), 54,8 (C), 61,7 (CH2), 80,8 (CH), 127,3 (CH), 128,6 (CH), 129,7 (C), 135,6 (C), 172,0 (CO), 174,3 (CO).
4.3.6 (2S*,3S*)-3-Benzyl-2-méthyl-5-oxotétrahydrofuran-3-carboxylate d’éthyle (cis–4c)
Huile. RMN 1H, δ (ppm) : 1,26 (t, 3 H, J = 7,1 Hz), 1,33 (d, 3 H, J = 6,6 Hz), 2,52 (d, 1 H, J = 17,3 Hz), 2,78 (d, 1 H, J = 13,7 Hz), 2,87 (d, 1 H, J = 17,3 Hz), 3,38 (d, 1 H, J = 13,7 Hz), 4,15–4,20 (m, 2 H), 4,51 (q, 1 H, J = 6,6 Hz), 7,02–7,10 (m, 2 H), 7,21–7,30 (m, 3 H). RMN 13C, δ (ppm) : 14,0 (CH3), 16,3 (CH3), 35,8 (CH2), 40,5 (CH2), 55,6 (C), 61,8 (CH2), 81,8 (CH), 127,4 (CH), 128,7 (CH), 129,8 (C), 135,5 (C), 171,3 (CO), 174,4 (CO).
4.4 Synthèse des bislactones 3. Procédure générale
Une solution de LHMDS (1,1 mmol ; 1,1 mL, 1 M dans le THF) est ajoutée goutte à goutte, sous N2 et à −78 °C, à une solution de lactone 4 (1 mmol) dans le THF (30 mL). Après agitation pendant une heure à −78 °C, l’aldéhyde (1,1 mmol) est ajouté et la solution est maintenue sous agitation à cette température pendant 1 h. Le milieu réactionnel est ensuite acidifié par addition d’une solution HCl dans MeOH. Après retour à 20 °C et addition d’eau (20 mL), le mélange est extrait à l’acétate d’éthyle (3 × 30 mL). Les phases organiques regroupées sont lavées à la saumure, puis séchées (MgSO4). Après évaporation des solvants sous pression réduite, le brut est fractionné ou purifié par chromatographie sur gel de silice (acétate d’éthyle/éther de pétrole, 20/80). Les bislactones 3 sont ensuite recristallisées dans un mélange hexane/toluène.
4.4.1 (3S*,3aR*,6aR*,6S*)-6-Ethyl-3a-méthyl-3-phényltétrahydrofuro[3,4-c]furan-1,4-dione (cis–3aa)
Cristaux blancs, F = 98–99 °C. RMN 1H, δ (ppm) : 1,01 (s, 3 H), 1,10 (t, 3 H J = 7,3 Hz), 1,71–1,92 (m, 2 H), 2,99 (d, 1 H, J = 2,4 Hz), 4,69–4,75 (m, 1 H), 5,71 (s, 1 H), 7,20–7,23 (m, 2 H), 7,38–7,42 (m, 3 H). RMN 13C, δ (ppm) : 9,7 (CH3), 19,6 (CH3), 29,4 (CH2), 51,5 (CH), 51,7 (C), 82,3 (CH), 85,2 (CH), 126,0 (CH), 128,9 (CH), 129,3 (CH), 134,8 (C), 175,3 (CO), 178,2 (CO). Analyse, calculé pour C15H16O4 : C 69,22 ; H 6,20 ; trouvé : C 69,07 ; H 5,95 %.
4.4.2 (3S*,3aR*,6aR*,6R*)-6-Ethyl-3a-méthyl-3-phényltétrahydrofuro[3,4-c]furan-1,4-dione (trans–3aa)
Solide amorphe. RMN 1H, δ (ppm) : 0,99 (s, 3 H), 1,15 (t, 3 H, J = 7,3 Hz), 1,76–2,15 (m, 2 H), 3,14 (d, 1 H, J = 6,6 Hz), 4,55–4,62 (m, 1 H), 5,70 (s, 1 H), 7,18–7,22 (m, 2 H), 7,38–7,41 (m, 3 H). RMN 13C, δ (ppm) : 10,6 (CH3), 17,3 (CH3), 24,1 (CH2), 48,6 (CH), 53,7 (C), 80,6 (CH), 84,3 (CH), 126,0 (CH), 129,0 (CH), 129,3 (CH), 135,1 (C), 172,5 (CO), 178,2 (CO). Analyse, calculé pour C15H16O4 : C 69,22 ; H 6,20 ; trouvé : C 69,16 ; H 6,13 %.
4.4.3 (3S*,3aR*,6aR*,6S*)-Hexyl-3a-méthyl-3-phényltétrahydrofuro[3,4-c]furan-1,4-dione (cis–3ab)
Cristaux blancs, F = 78–79 °C. RMN 1H, δ (ppm) : 0,88 (t, 3 H, J = 6,6 Hz), 1,01 (s, 3 H), 1,24–1,90 (m, 10 H), 2,98 (d, 1 H, J = 2,4 Hz), 4,76–4,82 (m, 1 H), 5,71 (s, 1 H), 7,20–7,24 (m, 2 H), 7,39–7,43 (m, 3 H). RMN 13C, δ (ppm) : 14,0 (CH3), 19,7 (CH3), 22,5 (CH2), 25,3 (CH2), 28,7 (CH2), 31,5 (CH2), 36,3 (CH2), 51,7 (CH), 51,7 (C), 81,1 (CH), 85,2 (CH), 126,0 (CH), 128,9 (CH), 129,3 (CH), 134,8 (C), 175,3 (CO), 178,2 (CO). Analyse, calculé pour C19H24O4 : C 72,13 ; H 7,65 ; trouvé : C 72,15 ; H 7,86 %.
4.4.4 (3S*,3aR*,6aR*,6R*)-6-Hexyl-3a-méthyl-3-phényltétrahydrofuro[3,4-c]furan-1,4-dione (trans–3ab)
Solide amorphe. RMN 1H, δ (ppm) : 0,90 (t, 3 H, J = 6,5 Hz), 1,00 (s, 3 H), 1,22–2,12 (m, 10 H), 3,15 (d, 1 H, J = 6,6 Hz), 4,64–4,70 (m, 1 H), 5,71 (s, 1 H), 7,20–7,23 (m, 2 H), 7,34–7,44 (m, 3 H). RMN 13C, δ (ppm) : 14,0 (CH3), 17,3 (CH3), 22,5 (CH2), 26,1 (CH2), 28,9 (CH2), 30,7 (CH2), 31,6 (CH2), 48,8 (CH), 53,5 (C), 79,3 (CH), 84,3 (CH), 126,0 (CH), 129,0 (CH), 129,3 (CH), 135,2 (C), 172,5 (CO), 178,2 (CO). Analyse, calculé pour C19H24O4 : C 72,13 ; H 7,65 ; trouvé : C 72,27 ; H 7,59 %.
4.4.5 (3S*,3aR*,6aR*,6S*)-3a-Méthyl-6-octyl-3-phényltétrahydrofuro[3,4-c]furan-1,4-dione (cis–3ac)
Cristaux blancs, F = 80–81 °C. RMN 1H, δ (ppm) : 0,89 (t, 3 H, J = 6,6 Hz), 1,03 (s, 3 H), 1,16–1,92 (m, 14 H), 2,99 (d, 1 H, J = 2,4 Hz), 4,78–4,83 (m, 1 H), 5,73 (s, 1 H), 7,22–7,27 (m, 2 H), 7,41–7,46 (m, 3 H). RMN 13C, δ (ppm) :: 14,1 (CH3), 19,7 (CH3), 22,6 (CH2), 25,3 (CH2), 29,0 (CH2), 29,1 (CH2), 29,3 (CH2), 31,8 (CH2), 36,3 (CH2), 51,7 (CH), 51,7 (C), 81,2 (CH), 85,2 (CH), 126,0 (CH), 128,9 (CH), 129,3 (CH), 134,8 (C), 175,3 (CO), 178,2 (CO). Analyse, calculé pour C21H28O4 : C 73,23 ; H 8,19 ; trouvé : C 71,97 ; H 8,33 %.
4.4.6 (3S*,3aR*,6aR*,6R*)-3a-Méthyl-6-octyl-3-phényltétrahydrofuro[3,4-c]furan-1,4-dione (trans–3ac)
Cristaux blancs, F = 98–99 °C. RMN 1H, δ (ppm) : 0,86 (t, 3 H, J = 6,6 Hz), 0,97 (s, 3 H), 1,20–1,60 (m, 12 H), 1,84–1,93 (m, 1 H), 1,98–2,08 (m, 1 H), 3,13 (d, 1 H, J = 6,6 Hz), 4,61–4,68 (m, 1 H), 5,69 (s, 1 H), 7,17–7,20 (m, 2 H), 7,38–7,41 (m, 3 H). RMN 13C, δ (ppm) : 14,1 (CH3), 17,3 (CH3), 22,6 (CH2), 26,2 (CH2), 29,2 (CH2), 29,3 (CH2), 29,4 (CH2), 30,7 (CH2), 31,8 (CH2), 48,8 (CH), 53,5 (C), 79,3 (CH), 84,3 (CH), 126,0 (CH), 129,0 (CH), 129,3 (CH), 135,1 (C), 172,5 (CO), 178,2 (CO). Analyse, calculé pour C21H28O4 : C 73,23 ; H 8,19 ; trouvé : C 73,46 ; H 8,48 %.
4.4.7 (3S*,3aR*,6aR*,6R*)-6-(4-Méthoxyphényl)-3a-méthyl-3-phényltétrahydrofuro[3,4-c]furan-1,4-dione (cis–3ad)
Cristaux blancs, F = 142–143 °C. RMN 1H, δ (ppm) : 0,84 (s, 3 H), 3,33 (d, 1 H, J = 2,0 Hz), 3,80 (s, 3 H), 5,78 (s, 1 H), 5,94 (s, 1 H), 6,93 (d, 2 H, J = 8,8 Hz), 7,21–7,24 (m, 2 H), 7,31 (d, 2 H, J = 8,8 Hz), 7,39–7,45 (m, 3 H). RMN 13C, δ (ppm) : 18,8 (CH3), 53,8 (CH), 55,3 (CH3), 80,3 (CH), 85,4 (CH), 114,4 (CH), 121,4 (C), 125,7 (CH), 126,0 (CH), 128,9 (CH), 129,3 (CH), 129,7 (C), 134,7 (C), 159,8 (C), 175,0 (CO), 178,4 (CO). Analyse, calculé pour C20H18O5 : C 71,00 ; H 5,36 ; trouvé : C 70,89 ; H 5,57 %.
4.4.8 (3S*,3aR*,6aR*,6S*)-3a-Benzyl-6-éthyl-3-phényltétrahydrofuro[3,4-c]furan-1,4-dione (cis–3ba)
Cristaux blancs, F = 160–162 °C. RMN 1H, δ (ppm) : 0,69 (t, 3 H, J = 7,1 Hz), 0,76–0,92 (m, 2 H), 2,08 (d, 1 H, J = 13,8 Hz), 2,83 (d, 1 H, J = 13,8 Hz), 3,09 (d, 1 H, J = 4,9 Hz), 4,48 (q, 1 H, J = 6,0 Hz), 5,81 (s, 1 H), 7,01–7,07 (m, 2 H), 7,24–7,27 (m, 3 H), 7,40–7,47 (m, 5 H). RMN 13C, δ (ppm) : 8,6 (CH3), 27,8 (CH2), 37,7 (CH2), 49,5 (CH), 58,5 (C), 81,4 (CH), 85,1 (CH), 126,0 (CH), 127,8 (CH), 128,9 (CH), 129,0 (CH), 129,2 (CH), 129,9 (CH), 134,1 (C), 135,2 (C), 174,7 (CO), 177,3 (CO). Analyse, calculé pour C21H20O4 : C 74,98 ; H 5,99 ; trouvé : C 75,19 ; H 5,90 %.
4.4.9 (3S*,3aR*,6aR*,6S*)-3a-Benzyl-6-hexyl-3-phényltétrahydrofuro[3,4-c]furan-1,4-dione (cis–3bb)
Cristaux blancs, F = 119–121 °C. RMN 1H, δ (ppm) : 0,66–0,77 (m, 2 H), 0,85 (t, 3 H, J = 7,1 Hz), 1,10–1,34 (m, 8 H), 2,06 (d, 1 H, J = 13,7 Hz), 2,80 (d, 1 H, J = 13,7 Hz), 3,08 (d, 1 H, J = 4,6 Hz), 4,54 (q, 1 H, J = 6,6 Hz), 5,80 (s, 1 H), 7,01–7,06 (m, 2 H), 7,23–7,33 (m, 3 H), 7,38–7,45 (m, 5 H). RMN 13C, δ (ppm) : 14,0 (CH3), 22,4 (CH2), 24,2 (CH2), 28,5 (CH2), 31,3 (CH2), 34,8 (CH2), 37,9 (CH2), 49,7 (CH), 58,4 (C), 80,6 (CH), 85,2 (CH), 126,0 (CH), 127,8 (CH), 128,9 (CH), 129,0 (CH), 129,2 (CH), 129,9 (CH), 134,3 (C), 135,3 (C), 174,9 (CO), 177,6 (CO). Analyse, calculé pour C25H28O4 : C 76,50 ; H 7,19 ; trouvé : C 76,26 ; H 7,43 %.
4.4.10 (3S*,3aR*,6aR*,6S*)-3a-Benzyl-6-octyl-3-phényltétrahydrofuro[3,4-c]furan-1,4-dione (cis–3bc)
Cristaux blancs, F = 85–87 °C. RMN 1H, δ (ppm) : 0,66–0,79 (m, 2 H), 0,88 (t, 3 H, J = 13,7 Hz), 1,20–1,36 (m, 12 H), 2,08 (d, 1 H, J = 13,7 Hz), 2,82 (d, 1 H, J = 13,7 Hz), 3,11 (d, 1 H, J = 4,6 Hz), 4,55 (q, 1 H, J = 6,6 Hz), 5,81 (s, 1 H), 7,01–7,06 (m, 2 H), 7,22–7,26 (m, 3 H), 7,39–7,45 (m, 5 H). RMN 13C, δ (ppm) : 14,1 (CH3), 22,6 (CH2), 24,2 (CH2), 28,9 (CH2), 29,1 (CH2), 29,2 (CH2), 31,8 (CH2), 34,8 (CH2), 37,9 (CH2), 49,7 (CH), 58,4 (C), 80,5 (CH), 85,2 (CH), 126,1 (CH), 127,8 (CH), 128,8 (CH), 129,0 (CH), 129,2 (CH), 129,9 (CH), 134,3 (C), 135,3 (C), 174,9 (CO), 177,5 (CO). Analyse, calculé pour C27H32O4 : C 77,11 ; H 7,67 ; trouvé : C 77,36 ; H 7,79 %.
4.4.11 (3S*,3aR*,6aR*,6R*)-3a-Benzyl-6-(4-méthoxyphényl)-3-phényltétrahydrofuro[3,4-c]furan-1,4-dione (cis–3bd)
Cristaux blancs, F = 200–202 °C. RMN 1H, δ (ppm) : 2,18 (d, 1 H, J = 13,8 Hz), 2,82 (d, 1 H, J = 13,8 Hz), 3,45 (d, 1 H, J = 6,3 Hz), 3,75 (s, 3 H), 5,53 (d, 1 H, J = 6,3 Hz), 5,91 (s, 1 H), 6,48 (d, 2 H, J = 8,8 Hz), 6,64 (d, 2 H, J = 8,8 Hz), 6,99–7,01 (m, 2 H), 7,17–7,26 (m, 3 H), 7,42–7,49 (m, 5 H). RMN 13C, δ (ppm) : 36,9 (CH2), 53,8 (CH), 55,3 (CH3), 59,0 (C), 79,7 (CH), 84,2 (CH), 114,2 (CH), 126,0 (CH), 126,6 (CH), 127,6 (CH), 128,8 (CH), 129,1 (CH), 129,2 (CH), 129,5 (C), 130,1 (CH), 133,6 (C), 134,8 (C), 159,9 (C), 173,7 (CO), 176,3 (CO). Analyse, calculé pour C26H22O5 : C 75,35 ; H 5,35 ; trouvé : C 75,08 ; H 5,33 %.
4.4.12 (3S*,3aR*,6aR*,6S*)-3a-Benzyl-3-méthyl-6-octyltétrahydrofuro[3,4-c]furan-1,4-dione (cis–3cc)
Cristaux blancs, F = 85–86 °C. RMN 1H, δ (ppm) : 0,68–0,79 (m, 2 H), 0,86 (t, 3 H, J = 7,1 Hz), 0,96–1,37 (m, 12 H), 1,56 (d, 3 H, J = 6,6 Hz), 2,75 (d, 1 H, J = 13,3 Hz), 2,96 (d, 1 H, J = 4,6 Hz), 3,35 (d, 1 H, J = 13,3 Hz), 4,42 (q, 1 H, J = 6,6 Hz), 4,85 (q, 1 H, J = 6,6 Hz), 7,11–7,19 (m, 2 H), 7,23–7,38 (m, 3 H). RMN 13C, δ (ppm) : 14,1 (CH3), 16,4 (CH3), 22,6 (CH2), 24,1 (CH2), 28,8 (CH2), 29,0 (CH2), 29,1 (CH2), 31,8 (CH2), 34,7 (CH2), 34,9 (CH2), 50,0 (CH), 57,5 (C), 80,3 (CH), 80,9 (CH), 128,0 (CH), 129,2 (CH), 130,1 (CH), 135,0 (C), 174,6 (CO), 177,2 (CO). Analyse, calculé pour C22H30O4 : C 73,71 ; H 8,44 ; trouvé : C 73,58 ; H 8,59 %.
4.4.13 (3S*,3aR*,6aR*,6R*)-3a-benzyl-6-(3,4,5-triméthoxyphényl)-3-méthyltétrahydrofuro[3,4-c]furan-1,4-dione (cis–3ce)
Cristaux blancs, F = 208–209 °C. RMN 1H, δ (ppm) : 1,60 (d, 3 H, J = 6,6 Hz), 2,74 (d, 1 H, J = 13,3 Hz), 3,36 (d, 1 H, J = 13,3 Hz), 3,37 (d, 1 H, J = 6,2 Hz), 3,66 (s, 6 H, 2 × CH3), 3,77 (s, 3 H), 4,95 (q, 1 H, J = 6,6 Hz), 5,42 (d, 1 H, J = 6,2 Hz), 5,98 (s, 2 H), 7,05–7,08 (m, 2 H), 7,16–7,18 (m, 3 H). RMN 13C, δ (ppm) : 15,7 (CH3), 34,3 (CH), 54,1 (CH3), 56,1 (2 × CH3), 57,9 (C), 60,7 (CH2), 79,3 (CH), 80,2 (CH), 101,4 (CH), 127,8 (CH), 128,9 (CH), 130,2 (CH), 130,2 (C), 132,9 (C), 134,3 (C), 153,4 (C), 173,5 (CO), 175,8 (CO). Analyse, calculé pour C23H24O7 : C 66,98 ; H 5,87 ; trouvé : C 66,70 ; H 5,72 %.
4.5 Synthèse de la bislactone 3af
4.5.1 Addition de l’éther iodométhylbenzylique sur les lactones trans–4a et cis–4a
Formation du dérivé iodé [11] : de l’iodotriméthylsilane (0,48 g, 2,43 mmol) est additionné, à 0 °C, sous N2 et sous agitation, à du di(benzyloxy)méthane (0,55 g, 2,43 mmol). Après agitation (30 min) à 0 °C, le mélange est dilué dans du THF (1 mL). Séparément, une solution de LHMDS (0,90 mL, 0,90 mmol, 1 M dans le THF) est ajoutée à une solution de lactone 4a (200 mg, 0,81 mmol) dans le THF (5 mL) à −78 °C. Après agitation à −78 °C (1 h), le dérivé iodé précédemment formé est transféré goutte à goutte à la seringue sur la solution contenant l’énolate. Le milieu réactionnel est maintenu sous agitation pendant 30 minutes supplémentaires à −78 °C puis ramené à température ambiante et neutralisé à l’aide d’une solution aqueuse saturée en NH4Cl. La phase aqueuse est ensuite extraite à Et2O (3 × 10 mL), les phases organiques regroupées sont lavées à l’eau (10 mL) et à la saumure (10 mL), puis séchées (Na2SO4). Après évaporation du solvant sous pression réduite, le résidu est purifié par chromatographie sur gel de silice (acétate d’éthyle/éther de pétrole, 20/80).
4.5.2 (2S*,3R*)-4-Benzyloxyméthyl-3-méthyl-5-oxo-2-phényltétrahydrofuran-3-carboxylate d’éthyle (trans–10a)
Au départ de trans–4a. Huile. RMN 1H, δ (ppm) : 1,02 (s, 3 H), 1,18 (t, 3 H, J = 7,2 Hz), 2,81 (t, 1 H, J = 3,9 Hz), 3,89 (d, 2 H, J = 3,9 Hz), 4,07–4,14 (m, 2 H), 4,42–4,52 (m, 2 H), 6,14 (s, 1 H), 7,26–7,43 (m, 10 H). RMN 13C, δ (ppm) : 14,0 (CH3), 21,3 (CH3), 52,2 (C), 52,8 (CH), 61,4 (CH2), 67,9 (CH2), 73,6 (CH2), 83,6 (CH), 126,5 (CH), 127,7 (CH), 127,8 (CH), 128,2 (CH), 128,3 (CH), 128,4 (CH), 135,4 (C), 137,3 (C), 172,2 (CO), 175,4 (CO). Analyse, calculé pour C22H24O5 : C 71,72 ; H 6,57 ; trouvé : C 72,01 ; H 6,38 %.
4.5.3 (2S*,3S*)-4-Benzyloxyméthyl-3-méthyl-5-oxo-2-phényltétrahydrofuran-3-carboxylate d’éthyle (cis–10a)
Au départ de cis–4a. Huile. RMN 1H, δ (ppm) : 0,88 (t, 3 H, J = 7,1 Hz), 1,54 (s, 3 H), 3,32–3,37 (m, 1 H), 3,62–3,75 (m, 2 H), 3,81 (dd, 1 H, J = 2,5, 9,9 Hz), 3,91 (dd, 1 H, J = 4,9, 9,9 Hz), 4,54 (s, 2 H), 5,38 (s, 1 H), 7,20–7,40 (m, 10 H). RMN 13C, δ (ppm) :: 13,5 (CH3), 16,1 (CH3), 48,3 (CH), 54,3 (C), 61,3 (CH2), 67,0 (CH2), 73,5 (CH2), 87,5 (CH), 125,6 (CH), 127,5 (CH), 127,8 (CH), 128,3 (CH), 128,5 (CH), 128,7 (CH), 135,2 (C), 137,5 (C), 172,0 (CO), 176,3 (CO). Analyse, calculé pour C22H24O5 : C 71,72 ; H 6,57 ; trouvé : C 71,70 ; H 6,66 %.
4.5.4 Hydrogénolyse des protections benzyliques
Une solution de lactone (trans–10a ou cis–10a) (0,5 mmol) dans EtOH (20 mL) en présence de Pd/C (5 mg, 10 %) est agitée sous atmosphère d’H2 (1 atm) pendant 12 h. Après filtration du catalyseur, le solvant est éliminé sous pression réduite et le résidu est purifié par chromatographie sur gel de silice (acétate d’éthyle/éther de pétrole, 40/60).
4.5.5 (2S*,3R*)-4-Hydroxyméthyl-3-méthyl-5-oxo-2-phényltétrahydrofuran-3-carboxylate d’éthyle (trans–11a)
Huile. RMN 1H, δ (ppm) : 0,96 (s, 3 H), 1,26 (t, 3 H, J = 7,1 Hz), 2,72 (t, 1 H, J = 4,6 Hz), 3,12–3,18 (m, 1 H), 3,97 (t, 2 H, J = 4,6 Hz), 4,22 (q, 2 H, J = 7,1 Hz), 6,03 (s, 1 H), 7,22–7,36 (m, 5 H). RMN 13C, δ (ppm) : 14,0 (CH3), 20,5 (CH3), 52,3 (C), 53,8 (CH), 60,2 (CH2), 61,9 (CH2), 84,0 (CH), 126,1 (CH), 128,4 (CH), 128,5 (CH), 135,1 (C), 172,6 (CO), 176,3 (CO). Analyse, calculé pour C15H18O5 : C 64,74 ; H 6,52 ; trouvé : C 64,49 ; H 6,22 %.
4.5.6 (2S*,3S*)-4-Hydroxyméthyl-3-méthyl-5-oxo-2-phényltétrahydrofuran-3-carboxylate d’éthyle (cis–11a)
Huile. RMN 1H, δ (ppm) : 0,91 (t, 3 H, J = 7,1 Hz), 1,58 (s, 3 H), 2,76–2,82 (m, 1 H), 3,40 (t, 1 H, J = 4,9 Hz), 3,65–3,78 (m, 2 H), 3,92–4,04 (m, 2 H), 5,34 (s, 1 H), 7,20–7,36 (m, 5 H). RMN 13C, δ (ppm) : 13,5 (CH3), 17,4 (CH3), 48,4 (CH), 54,3 (C), 59,3 (CH2), 61,5 (CH2), 87,6 (CH), 125,8 (CH), 128,3 (CH), 128,9 (CH), 135,0 (C), 171,8 (CO), 177,1 (CO). Analyse, calculé pour C15H18O5 : C 64,74 ; H 6,52 ; trouvé : C 64,67 ; H 6,78 %.
4.5.7 Traitement des alcools en présence d’acide
Une solution d’alcool (trans–11a ou cis–11a) (0,5 mmol) et d’acide para-toluènesulfonique (APTS, 1–2 mg) dans le toluène (20 mL) est portée à reflux pendant quatre heures. Après refroidissement, le solvant est éliminé sous pression réduite et le résidu est purifié par chromatographie sur gel de silice (acétate d’éthyle/éther de pétrole, 20/80).
4.5.8 (3S*,3aR*,6aR*)-3a-Méthyl-3-phényltétrahydrofuro[3,4-c]furan-1,4-dione (3af)
Au départ de trans–11a. Cristaux blancs, F = 137–138 °C. RMN 1H, δ (ppm) : 1,00 (s, 3 H), 3,20 (d, 1 H, J = 6,7 Hz), 4,51 (dd, 1 H, J = 6,7, 9,5 Hz), 4,77 (d, 1 H, J = 9,5 Hz), 5,75 (s, 1 H), 7,19–7,22 (m, 2 H), 7,41–7,43 (m, 3 H). RMN 13C, δ (ppm) :: 17,4 (CH3), 46,3 (CH), 51,3 (C), 67,7 (CH2), 85,3 (CH), 126,0 (CH), 129,1 (CH), 129,5 (CH), 134,9 (C), 175,1 (CO), 178,9 (CO). Analyse, calculé pour C13H12O4 : C 67,23 ; H 5,21 ; trouvé : C 66,98 ; H 5,38 %.
4.5.9 (2S*,3S*)-3-Méthyl-4-méthylèn-5-oxo-2-phényltétrahydrofuran-3-carboxylate d’éthyle (12)
Au départ de cis–11a. Huile. RMN 1H, δ (ppm) : 0,88 (t, 3 H, J = 7,1 Hz), 1,64 (s, 3 H), 3,56–3,74 (m, 2 H), 5,20 (s, 1 H), 5,66 (s, 1 H), 6,35 (s, 1 H), 7,29 (s, 5 H). RMN 13C, δ (ppm) :: 13,4 (CH3), 20,9 (CH3), 54,7 (C), 61,3 (CH2), 85,9 (CH), 122,4 (CH2), 125,6 (CH), 128,2 (CH), 128,8 (CH), 134,8 (C), 140,2 (C), 168,9 (CO), 170,1 (CO). Analyse, calculé pour C15H16O4 : C 69,22 ; H 6,20 ; trouvé : C 69,05 ; H 6,51 %.
CCDC 811701 contient les détails supplémentaires de la résolution structurale pour cet article. Ils peuvent être obtenus sans frais sur demande au Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/conts/retrieving.html.
Remerciements
Ce travail a bénéficié de l’aide financière de ADIR (laboratoires Servier) et du programme PRIM (région Nord-Pas-de-Calais). Les auteurs tiennent à remercier le Pr. G. Nowogrocki (UCCS-UMR 8181-UST Lille 1) pour la détermination de la structure cristalline et le Pr. E. Deniau pour ses commentaires avisés.